Researchers have been trying to find a full definition of aromaticity for almost two centuries, and yet keep discovering new types. Katrina Krämer asks when chemists will get to the bottom of this elusive concept
‘Controversial’, ‘suspicious’, ‘questionable’ and even ‘an exercise in chemical futility’ – these are only some of the ways scientists have described the concept of aromaticity. And that’s in spite of it being a concept that almost all chemists are familiar with from school or university.
‘There are a lot of mixed feelings about the term aromaticity,’ says computational organic chemist Judy Wu from the University of Houston, US. ‘Some people find it to be so fuzzy that it’s not serious science. But I think that’s what makes it even more remarkable – that something that feels so fuzzy actually can give useful explanations for chemistry.’
PubChem, the largest public database of chemical compounds, contains almost 110 million structures. There’s an estimate that two-thirds of them are fully or partially aromatic. Understanding how aromaticity influences a compound’s properties allows researchers to design everything from drugs to molecular light emitters. ‘I think aromaticity is one of the concepts that really connects to the most important properties in chemistry,’ says Mercedes Alonso, a quantum chemist at the Vrije Universiteit Brussel in Belgium. ‘It’s not only an electronic property, it will influence the structure, it will influence the supramolecular arrangements that the compounds make, the biological activity.’
The problem with aromaticity is that, despite 200 years of research, there’s no clear definition, rules or experiments that can identify it among all compounds. Benzene might look very different from a heptaborate polyhedron B7H72−, the aluminium square Al42– or a gigantic molecular wheel made of more than 1000 carbon atoms – yet they are all considered aromatic by one standard or another.
‘It’s a paradox,’ says Renana Poranne who researches computational physical organic chemistry at ETH Zürich in Switzerland. ‘On the one hand, [aromaticity] is very ill defined, and on the other hand apparently quite useful for identification or having some expectations about the properties of a chemical compound.’ But this is part of what fascinates researchers. ‘People are interested to understand aromaticity because we still don’t,’ she says.
Despite chemists having access to an ever-increasing number of shorthand rules and a vast array of computational methods, they keep finding aromaticity in unexpected places. But with more than 20 different types of aromaticity described so far, how close are chemists to finding a definition that unites all of them?
Back to the textbooks
Many chemists first come across the concept of aromaticity in their undergraduate textbook or first-year lectures. According to these, all that’s needed to identify aromaticity are four easy rules: a molecule must be cyclic, flat, have a continuous ring of p-orbitals and 4n + 2 π-electrons (with n being an integer).
The last one, Hückel’s rule, is probably the most memorised of them all. It was developed (if not explicitly stated) by German physical chemist Erich Hückel in 1931 as part of the simplified quantum-mechanical calculations he used to deal with unsaturated organic molecules. He noticed that having 4n + 2 π-electrons gave aromatic molecules a filled π molecular orbital shell, equivalent to the filled atomic orbital shell that gives noble gases their extraordinary stability. Hückel’s rule remained obscure until almost two decades later, when chemists first used it to validate tropone’s aromaticity.
There is no ’magic’ in having six π electrons
But over the course of the 20th and 21st centuries, scientists discovered that these rules don’t apply for all but a small subset of aromatics. In fact, the most archetypal of all aromatics – benzene – ‘is not representative of the majority of aromatic molecules’, wrote Wu.
There is a whole range of aromatics that aren’t flat – boron–hydrogen clusters, for example, as well as fullerenes (although the aromaticity of buckminsterfullerene C60 is still under debate, with various researchers claiming it to be aromatic, antiaromatic and non-aromatic). And the members of the σ and δ aromaticity families add to the debate over whether aromaticity is only for molecules with p orbitals. The tritantalum oxide cluster Ta3O3– contains delocalised electrons in d orbitals, whereas the trihydrogen cation’s (H3+) aromatic properties are based on the delocalisation of s orbital electrons.
On the opposite end of the spectrum, there are molecules that look aromatic but are not. Borazine, a six-membered ring made up of alternating boron and nitrogen atoms, is sometimes called ‘inorganic benzene’. It has six π electrons and follows all other aromaticity rules. ‘Yet it turns out it doesn’t actually have an appreciable π current, because of the electronegativity and -positivity of the boron and nitrogen atoms,’ explains Poranne. Borazine is most likely to be only very moderately aromatic. This goes to show that ‘there is no “magic” in having six π electrons’, wrote Wu.
Not according to rules
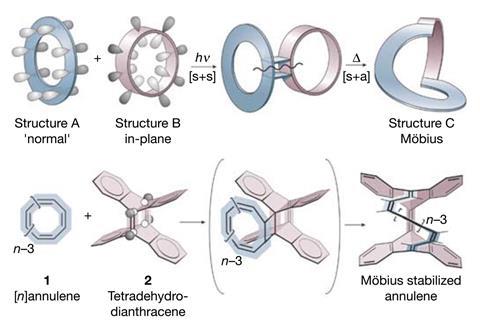
There are now at least another 18 sets of rules in addition to Hückel’s. Many of them require additional information about a molecule’s structure to not seem contradictory to other rules. For example, while Hückel aromatics have 4n + 2 π-electrons, those with a half-twist Möbius structure need 4n π-electrons to be aromatic.
‘The [definition] that most people agree with is the one by [Paul von Ragué] Schleyer in his Chemical Reviews paper in 2005,’ says computational chemist Miquel Solà from Spain’s University of Girona. Schleyer and his team wrote that aromaticity is a manifestation of electron delocalisation in closed circuits – and that all the different tests and rules simply focus on the results of the fact. There are four main criteria that, when combined, may offer some insight into a compound’s aromaticity: structure, reactivity, energy and magnetic properties.
Many aromatics are planar and have equal bond lengths. Benzene’s six carbon–carbon bonds are all equal length compared with those calculated for cyclohexatriene, a non-existent model molecule with fixed double bonds that are shorter than its alternating single bonds. But on its own, this criterion is not much use. There are linear compounds like the polymethinium ion C5H9N2+ that have almost equal carbon–carbon bond length despite not being aromatic. And there’s a whole class of decidedly non-planar 3D aromatics.
Equally, reactivity is a minefield of exceptions. Benzene undergoes electrophilic aromatic substitutions rather than addition reactions, but even simple polycyclic compounds like anthracene prefer additions despite being aromatic.
We have more than 100 descriptors to quantify aromaticity, but there is not a single one that can be applied universally
Aromaticity is associated with stability. Benzene is often quoted to have a 30kcal/mol stabilisation energy because of its aromaticity. But what the textbooks then fail to mention is that this varies hugely depending on which reference you look at. While aromatic compounds should be more stable than their non-aromatic counterparts, it’s often impossible to find a non-aromatic molecule that closely resembles the aromatic in geometry and electronic features.
In the 1930s, two teams deduced benzene’s resonance energy in comparison to polyenes and cyclohexene, respectively. They both arrived at a value of around 36kcal/mol. But in 2007, Schleyer’s team found that when correcting for hyperconjugation, benzene’s resonance energy almost doubles.
The problem with aromaticity, says Alonso, is that it’s ‘a concept that we have invented for trying to describe the properties of a compound’. Unlike a melting point or molecular mass, aromaticity is not an observable property that can be measured in the lab – a fact that seems unpalatable to many scientists. ‘We have more than 100 descriptors to quantify aromaticity, but there is not a single one that can be applied universally,’ explains Alonso.
Mostly magnetic
Over the last few decades, examining a molecule’s magnetic properties has become the most widespread way to describe aromaticity. ‘Nics is definitely the most popular aromaticity criterion out there today,’ says Poranne.
The nucleus independent chemical shift (Nics) was developed in 1996 by Schleyer (although Michael Bühl and Christoph van Wüllen calculated essentially the same thing a year earlier without naming it) and chemists started using his ‘simple and efficient aromaticity probe’ enthusiastically. Schleyer’s original article has amassed more than 4000 citations – for good reason, says Poranne. ‘It’s just super-easy to use, and it is implemented in the most commonly used computational software.’
Nics is a cleverly designed aromaticity probe based on nuclear magnetic resonance (NMR) – another reason it is popular with experimentalists. When aromatics are placed in a magnetic field, like the one inside an NMR machine, its π electrons start to race around the ring – the aromatic ring current. This electron movement, in turn, creates its own magnetic field perpendicular to the ring. In benzene, this bagel-shaped magnetic field becomes visible as a dramatic influence on the hydrogen atoms’ NMR chemical shifts.
The reason chemists don’t just use spectra to determine aromaticity is because there is a huge group of molecules that can’t simply be stuck inside an NMR machine. They might be too unstable or simply don’t have the hydrogen or carbon atoms necessary. And often, the spectra of larger polycyclic aromatic hydrocarbons can get exceedingly difficult to read as signals crowd around the 8ppm mark.
Moreover, atoms are influenced by things other than just aromaticity. So Nics doesn’t look at the chemical shift of a real atom, it uses a ghost atom. Named bq after the ghostly Lord Banquo from Shakespeare’s Macbeth, it has no protons, neutrons or electrons to disturb the measurements. Bq is merely a point in space, placed in the middle of the ring to act as a sensor for the magnetic environment around it. ‘The computation then asks the question: “What is the chemical shift at that point in space?”,’ explains Poranne.
Nics’ single-value answer not only indicates whether something is aromatic or not, but also how strongly so. The more negative the value, the stronger its aromaticity. Values hovering around 0 are for non-aromatics whereas strongly positive values indicate antiaromaticity.
The problem with Nics
But soon after Schleyer’s initial proposal, researchers became worried about Nics’ many pitfalls – such as disturbances from σ orbitals or other non-aromatic magnetic field lines. Some argued that Nics, as a virtual, non-testable parameter, can’t be used to assess a chemical fact and should be abandoned.
Some chemists started to refine Nics to iron out its problems, which is why there are now many different Nics versions out there. Others devised alternative methods to assess aromaticity’s magnetic criterion. For example, Dage Sundholm’s Arcs (aromatic ring current shieldings) measure induced ring current strength, which allows for better comparisons between unrelated molecules than Nics does. And Paolo Lazzeretti’s polygonal current model (PCM) can deal with molecules’ actual geometric structure better than Nics, making more accurate evaluations for small systems such as the σ aromatic cyclopropane and the π aromatic tetra-azadianaion N42−.
Sundholm and his team used Arcs and another of their methods – Gimic or gauge-including magnetically induced current – to disprove so-called hydrogen-bonded aromaticity in the hydrogen fluoride trimer (HF)3. Despite its ring shape, planarity and surprisingly negative Nics value, Gimic showed its ring current to be too weak to be called aromatic. The Nics value was simply a result of strong edge currents circling around each HF monomer rather than an aromatic ring current, Sundholm and colleagues wrote.
Nevertheless, Nics became – and remains – hugely popular to categorise aromaticity, although in many instances it doesn’t just struggle with ‘unusual’ aromatics but also with classic organic ones. One such case is polycyclic aromatic hydrocarbons, systems in which individual rings can be anti-aromatic while simultaneously participating in a semi-local or global aromaticity.
Anthracene, for example, is made up of three linearly fused benzene rings. Nics shows the middle one to have the most negative value. ‘If you were just interpreting it at face value, you would say that means the middle ring is the most aromatic,’ says Poranne. ‘So why is the most aromatic [ring] the most reactive? We assume that most aromatic [part] should be most stable.’
In such cases, she points out, it might not make sense to talk about the rings as separate units when they are in fact a fully conjugated system. Although Nics is calculating the induced magnetic field in each ring, it’s not measuring one ring current at a time.
On its own, Nics is clearly not sufficient to declare aromaticity, Alonso says. ‘Imagine that a compound only fulfils the magnetic criteria, but not the energetic one – in my view, this compound cannot be described as fully aromatic.’
Aromaticity in abundance
With Nics as well as numerous experimental and computational methods in hand, chemists discovered several new types of aromaticity around the turn of the millennium, including spherical aromaticity in fullerenes and the first Möbius aromatic molecule in 2003. But the one that surprised chemists the most might be the first all-metal aromatic in 1995.

Gregory Robinson, who had been working with gallium compounds for several years, discovered a cyclogallane with a triangular Ga32– core. The triangle followed all of the classic aromaticity markers including planarity, electron delocalisation, bond length equalisation as well as the Hückel rule with its two π electrons. Robinson proposed the cyclogallane to be isoelectronic with the aromatic cyclopropenium cation. Only one year later, his team confirmed their hypothesis using the newly minted Nics. ‘I think this was the starting point for thinking differently about aromaticity,’ says inorganic chemist Stefanie Dehnen from the University of Marburg in Germany.
A few years later, teams led by experimental physical chemist Lai-Sheng Wang from Brown University in Rhode Island, US, and computational chemist Alexander Boldyrev claimed the discovery of a second π aromatic metal cluster, Al42–. Wang recalls that it was initially difficult to convince the community of their findings. One of the reasons for this was that aromaticity assessments rely heavily on computational means. ‘A lot [of experimental chemists] were quite sceptical about computational chemistry, because of the accuracy at the time was, admittedly, limited,’ he says. Nevertheless, most turned around eventually.
Not so theoretical chemist and 1981 Nobel prize laureate Roald Hoffmann. In 2015, he wrote that while he didn’t doubt that Al42– was a real molecule, ‘what grates on me are claims of aromaticity … I would be willing to bet a good bottle of NY State Riesling (I will leave a sum for the eventuality in my will) that salts of these will not be made in milligram quantities in my lifetime or yours.’
What we are missing is a unified theory of aromaticity
‘He argued that these [clusters] are only gas phase molecules, he wanted something that you could put in a bottle,’ Wang says. After all, aromaticity is supposed to impart stability. ‘But there are a lot of molecules in outer space, it’s all in the gas phase. For [gas phase] clusters, it’s more difficult to really pin down their reactivity. But there’s a certain electronic stability to the electron delocalisation, there’s no question about that.’
Hoffmann felt people might be overusing the word aromaticity, stretching the definition too thin to still be useful. ‘In some sense, I can agree with him,’ says Wang. A single calculated value on its own does little to confirm a compound’s aromaticity. ‘Because yes, a lot of people just do a calculation to say this is aromaticity or that is. Some of these may not have any substance to them.’ This might for example be the case when a molecule’s most stable structure is unclear.
While Alonso doesn’t agree with all of Hoffmann’s points, she too thinks that there might be a danger of overusing the concept. ‘What we are missing is a unified theory of aromaticity, something most of the people that are working on aromaticity can feel comfortable with,’ she says.
All the way from p to s
After first revealing the aromatic nature of some metal clusters, researchers were eager to dig deeper, finding many that they claimed were not only π but also σ aromatic. Some compounds, like Wang and colleagues’ aluminium cluster, even showed both types of aromaticity, though this remains heavily debated. ‘Nics and Gimic calculations do not agree in the type of electrons that cause the ring current,’ with the former pointing towards σ and π contributions and the latter towards mostly σ, explains Florian Weigend, a quantum chemist at the University of Marburg.
But in clusters, valence electrons delocalise over the entire molecule, which doesn’t necessarily make them σ aromatic. ‘Along the metal–metal interaction axis there should be some kind of electron dynamics more so than in carbon compounds with defined covalent bonds,’ Dehnen explains. When non-localisable electron density was first observed in boron cages, it was a spectacular discovery, so it is understandable why the term 3D σ aromaticity was created, she says. ‘Of course, there is σ aromaticity, but don’t call everything made of metal atoms that has delocalised orbitals σ aromatic.’
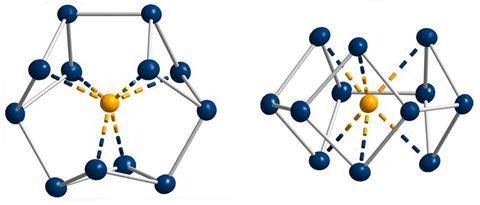
In 2020, her team and that of Florian Weigend synthesised a bismuth–thorium cluster, the biggest π aromatic molecule made purely from metal atoms. ‘I’d say the term aromaticity should be resharpened a bit, and really reserved for these very unusual things,’ Dehnen says. ‘In organic chemistry, it’s the delocalisation itself, and in other areas like metal clusters or inorganic compounds it should be reserved to π systems. I think this sticks more to the original definition.’
But as Poranne and Amnon Stanger wrote in 2015: ‘In many ways, aromaticity is like beauty, namely, in the eye of the beholder.’ ‘There’s no right or wrong, it’s about explaining what we see from two sides,’ says inorganic chemist Zhong-Ming Sun from Nankai University in China. His team, together with those of Wang and Boldyrev, isolated an all-metal antiaromatic in the solid state in 2016. Since electrons in such clusters are more delocalised and prone to form multi-centre bonds than those in organic compounds, ‘you can explain [the bonding] using antiaromaticity, but you can also explain them using traditional chemical bonding concepts,’ Sun says. He hopes that making more and different clusters might provide more evidence to support one or the other view.
‘In my opinion, it’s not only π aromaticity, you have system with σ, π, δ and maybe φ aromaticity,’ says Solà. ‘If you talk to people working in the field, the name that they use is superatoms for clusters that are closed shell,’ he explains. ‘But when we look at the parameters related to aromaticity, we conclude that these systems have aromatic properties. So we prefer to call them aromatic.’
With so many computational methods available, chemists can amass huge amounts of data for every compound. ‘But it’s not always easy to interpret it,’ says Poranne. ‘We are left with a lot of overinterpretation or hand-waving results and discussion of what the results mean. One of the things that we’re working on is how do you take all of this data and really make it into one interpretable picture of how this relates to what this molecule is expected to do.’
‘[Chemists] are starting to realise that there is some chemical power that we can harness from this simple concept,’ says Wu.
More than just an academic argument
‘What we are doing is trying to find connections between aromaticity and molecular properties that have real-world meaning, like Homo–Lumo gap or ionisation potential, which are important for functional materials,’ says Poranne. In 2018, she discovered that by defining a few simple building blocks and combination rules, she could predict the shape of Nics scans in polycyclic aromatic hydrocarbons. She and her team have now expanded this method to predict what happens to these molecules in the excited state when aromatics become antiaromatics – and vice versa.
Alonso and her team found that certain porphyrin-type molecules can switch between aromatic and antiaromatic Hückel as well as Möbius aromatic structures. Since each of these forms has different conductivity, they could find use as switches in single molecule electronics and thermoelectric devices.
We’ve evolved past the stage of asking what aromaticity means
Wu and her team showed that aromaticity even plays a role in the human body, as a driving force for DNA base pairing and DNA’s photostability. ‘We’ve translated what is happening [in DNA photodeactivation] into a useful chemical language,’ she explains. While the process could also be explained with physical terms like ionisation potential, ‘you cannot go to an organic chemist and say: “Can you please design a hydrogen-bonded system with a high ionisation energy here and a low ionisation energy there.” But aromaticity and antiaromaticity is very closely tied to a chemical structure, [it’s] a guideline for designing a system that does what we want.’
With aromaticity now firmly past being purely an academic argument, is overhauling the definition even needed? Solà thinks so, particularly when it comes to borderline systems like metal clusters. In 2018, he closely examined 19 aromaticity rules, taking the first steps to combine and connect them with the eventual goal to find a single aromaticity theory that unifies them all.
Sun thinks that there eventually will be an update to include these systems, but that, for now, chemist will have to describe new concepts using old words – despite the issues this sometimes causes. ‘I don’t think we will ever get to a time where everybody agrees there’s one correct method that we should all use exclusively,’ says Poranne. ‘I don’t think that exists in science in general.’
‘I think as the field moves forward, the types of questions that are relevant also are evolving,’ says Wu. ‘We’ve evolved past the stage of asking what aromaticity means. Now is the time to ask the question “What do we use it for?”’
Katrina Krämer is a science correspondent for Chemistry World









No comments yet