Computational fitting procedure set to reinvigorate vibrational circular dichroism spectroscopy
A process to improve the reliability of a technique that can determine a molecule’s chirality could see the tool applied on a bigger scale. The process accounts for uncertainties in the theoretical energies of conformers of chiral molecules, enabling a more reliable fitting of experimental vibrational circular dichroism (VCD) spectra by the calculated spectral contributions of individual conformers.
The ability to distinguish between enantiomers is an important consideration when working with chiral molecules, as their properties can be very different. Take the molecule carvone: its two enantiomers interact differently with olfactory receptors so a discerning nose can determine which is which – one smells of spearmint, the other of caraway seeds. However, the presence of different enantiomers can be far more difficult to detect and have far greater consequences.
The most well established technique for this task relies on the difference in circular dichroism (see box below) between enantiomers as mirror image molecules absorb the two different types of circularly polarised light (right- and left-handed) to different extents. Measuring that difference across a wavelength range produces a spectrum, from which scientists can begin to understand which enantiomers are present. As the spectrum alone does not provide sufficient information for enantiomeric determination, computational modelling interprets the experimental data by comparing it with theoretically predicted spectra.
The beauty of our approach is that the correct handedness always gives better agreement with the experiment than the opposite handedness
Vibrational circular dichroism spectroscopy, however, is very sensitive to molecular conformation, and, at room temperature, large and flexible molecules exist in many conformations. Each structure has its own circular dichroism fingerprint, and thus an experimental spectrum is actually a superposition of the spectra of many individual conformers in the sample. Conformers do not occur in equal amounts, either – the more stable, low energy structures will be more common than higher energy alternatives. To take account of this, it is customary to determine the possible conformations of a molecule, calculate their energies and corresponding fingerprints, and then average these individual components to fit the experimental VCD spectrum.
This is not the end of the story. ‘Calculations can be done with various methods and at different levels, from very simple to very advanced. Each of these calculations leads to different relative energies of the possible spatial structures, and thus ultimately to a different theoretical spectrum that is subsequently compared with the experimental one – which at the end of the day is the basis for the assignment of their handedness,’ explains Wybren Jan Buma, from the University of Amsterdam, the Netherlands.
By using a genetic algorithm that can account for uncertainties in calculated energies, Buma and his colleagues have produced a statistically-justified method for fitting calculated spectra to experimental VCD spectra. This survival-of-the-fittest approach to obtaining the optimum experimental fit aims to reduce the chance of an incorrect conclusion regarding which enantiomer is present.
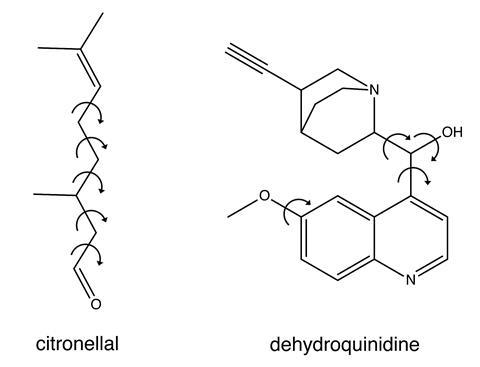
Buma’s team tested the process on two molecules, citronellal and dehydroquinidine. Citronellal is an acyclic monoterpene with insect repellent properties and the five carbon–carbon single bonds along its backbone mean it has an extremely flexible structure. Dehydroquinidine is a type of chincona alkaloid whose conformational landscape is known to be the dominant factor in determining the outcome of asymmetric organocatalysis reactions. The team showed that even for cases exhibiting large dynamic structural changes, their process is useful in distinguishing the correct absolute configurational assignment. Dehydroquinidine served as a worst-case test of the process as it exhibits large dynamic structural changes around its polar hydroxyl group, the type of which have resulted in conflicting conclusions in previous VCD studies. ‘The beauty of our approach is that the correct handedness always gives better agreement with the experiment than the opposite handedness,’ says Buma. ‘And, even more importantly, we can give a quantitative measure of how reliable the assignment is.’
Automation ambitions
Buma’s team hope their process will improve the reliability of VCD as an analytical tool, and result in its use becoming more widespread. ‘We will be looking at different systems, for example self-assembling polymers, fibrils, amyloids … which at this moment are very difficult to handle. One of the other things we are trying to do is incorporate all of these ideas into a kind of [automated] mainframe, like those for NMR.’
According to Prasad Polavarapu, an expert in chiroptical spectroscopic methods at Vanderbilt University, US, the process represents a useful advance. ‘However, the major problem of independently confirming the conformer populations deduced in this manner with experimentally determined conformer populations in solution phase will haunt the researchers for years to come. This is because the information on experimentally determined conformer populations in solution phase is rather sparse,’ he adds.
Petr Bour, who has used VCD to probe the structure of helical molecules at the Czech Academy of Sciences in Prague, says the process provides more information about molecular behaviour. ‘I see some dangers in the approach related to the possible over-fitting and numerical instabilities of the results, but in general I agree that with their method VCD spectroscopy becomes a robust tool to study chiral molecules.’
Circular dichroism
For transverse waves, such as light, the direction of the oscillation of the wave is perpendicular to the direction the wave is travelling in. These waves have a property called polarisation, which determines the orientation of these oscillations. For electromagnetic waves such as light, this refers to the electric field component of the wave. The electric field can oscillate vertically, horizontally, or at any angle perpendicular to the direction of travel, and one can express all polarised light states as a sum of two states of vertically and horizontally polarised light. If these two polarised states are out of phase with each other by a quarter of a wavelength, the overall polarised state will cease to be linear (that is, not confined to one plane), and the resultant oscillation will rotate around the direction of travel at a constant rate and amplitude. This type of light is known as circularly polarised light, and it comes in two types; right- and left-handed. The difference between these two is whether the rotation of the oscillation is clockwise or anti-clockwise, respectively.
Optically active molecules absorb these two types of circularly polarised light to different extents, and as such, one can measure the different in absorption between the two types – this is referred to as circular dichroism. Measuring the circular dichroism over a range of wavelengths is the process known as circular dichroism spectroscopy, and it is particularly useful in elucidating details of secondary structure and conformations of large biomolecules, many of which are chiral. Vibrational circular dichroism spectroscopy measures circular dichroism over energy ranges relevant to vibrational motion; namely, the infrared and near-infrared section of the electromagnetic spectrum.











No comments yet