A team of scientists based in Sweden and the UK has developed a synthetic screening method that uses stopped-flow chemistry and machine learning to accelerate drug discovery through diversity-oriented synthesis.1

Stopped-flow chemistry is an alternative method to traditional batch and continuous flow processes. By arresting the flow of reacting materials within the system, the platform allows high-speed reactions to be performed under superheated and high-pressure conditions in flow, with easily varied conditions. Compared to other methods, stopped-flow chemistry requires minimal reagent quantities and solvent volumes, does not depend on the flow rate used, and allows users to construct large libraries of chemicals rapidly and easily. ‘The integration of a stopped‑flow reactor with robotic liquid handling units enables facile access to a diverse library of starting reagents while reducing the dead time and waste generation during experimentation,’ explains Milad Abolhasani, an expert in flow chemistry from NC State University, US, who wasn’t involved in the study.
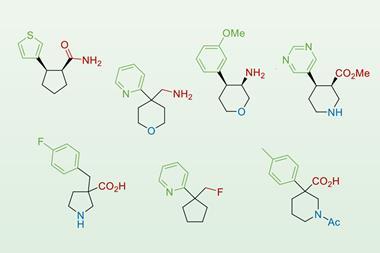
Amide bonds are ubiquitous in active pharmaceutical ingredients and making them accounts for about 16% of reactions in medicinal chemistry laboratories.2 Despite this, reaction success is variable, and while parallel batch setups can react many starting material combinations at once, a success rate of 50–80% is still typical for a diverse library. However, a team based between the University of Leeds and AstraZeneca has now shown that merging predictive computational tools with stopped-flow chemistry can raise the success of library syntheses to 100%.
The scientists first optimised the temperature and reaction time conditions for two combinations of carboxylic acid, amine and coupling agent materials. To simplify the analysis, the team combined various temperature and reaction time parameters and ranked them on a nine-point scale from mild to harsh.
They then reacted five sterically hindered acids and five electronically-unfavoured amines to make 25 distinct amide products. Every reaction was performed using each of the nine points on the reaction parameters scale, in each case using one of four coupling reagents, generating data for 900 individual reactions in 192 hours.
The scientists successfully synthesised all desired products and identified the optimal conditions in each reaction. They also used 90% less starting material than a traditional continuous flow approach. Team leader Richard Bourne says their method ‘provides much more accurate data than well plate-based chemistry, which has much poorer heat transfer and the potential for degradation as the samples are not directly analysed after each individual reaction is completed.’
Using an unoptimised set of temperature and reaction time conditions across all the reactant combinations by traditional methods would have resulted in a success rate as low as 16%, the team estimates. The stopped-flow approach therefore allowed them access to otherwise challenging chemical space. According to Abolhasani, ‘to accelerate the drug discovery timeframe, [researchers] need to have access to a larger part of the experimental space with high data reproducibility. This work is a great effort toward unlocking access to these unexplored spaces in the early stages of drug discovery.’
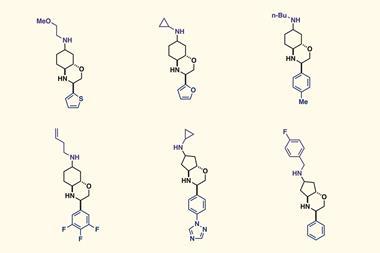
The researchers used their optimisation data within a predictive tool to make a 30-product amide library. They found that a machine learning model based on these results could predict optimal conditions for this new set of reactions with 92% accuracy. As a result, they hope that similar work will enhance the predictability of synthetic processes for diversity-oriented synthesis within drug discovery. Bourne says ‘in around 10 years I hope that we will be at a point where we can accurately predict reaction conditions for specific substrates with only minimal new experimental training datasets required.’
References
These articles are open access
1 C Avila et al, Chem. Sci., 2022, DOI: 10.1039/d2sc03016k
2 A I Alfano, H Lange and M Brindisi, ChemSusChem, 2022, 15, e202102708 (DOI: 10.1002/cssc.202102708)










No comments yet