Catalyst’s carbene pocket shows a preference for specific links without needing a directing group
Chemists in the US have designed a catalyst that can activate C–H bonds selectively, without the need for the presence of a more reactive functional group or the use of external directing groups. The finding opens the way to more efficient syntheses of complex organic molecules from simpler precursors.
Because C–H bonds are relatively inert, and because there are typically multiple C–H bonds in a given organic molecule, targeting a specific bond and functionalising it is inherently tricky. A well-established technique is to use directing groups, which direct the reaction to a particular bond, after which the directing group is removed. This, however, adds extra steps to the process and requires stoichiometric quantities of the directing group.
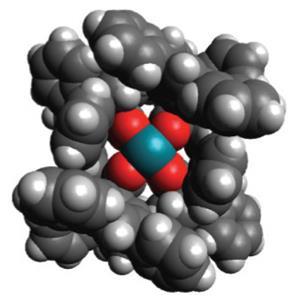
Now, a team at Emory University in Atlanta, led by Huw Davies, has shown that a catalyst can achieve site-selective and stereoselective functionalisation of unactivated C–H bonds. The key to the system is a catalyst that is highly sterically demanding: it will accept the C–H bond from the substrate only if that bond has the appropriate stereochemical ‘credentials’.
The researchers designed a catalyst that consists of a dirhodium centre encased by a bulky organic scaffold. The architecture of the catalyst dictates which C–H bond of the substrate will be able to occupy its active pocket.
To demonstrate the concept, the team used n-pentane as a model. This contains three types of C–H bond that would normally be considered unreactive and difficult to distinguish during a chemical transformation. ‘From purely steric considerations the C-1 carbon of the pentane would be expected to react preferentially,’ Davies explains. ‘However, electronically the C-2 and C-3 carbons would be preferred.’
The catalyst is first presented with a diazo compound which is readily decomposed by the rhodium to generate a highly reactive ‘donor–acceptor’ carbene, which remains clamped within the catalytic pocket. ‘Electronically these carbenes have a preference to react with the more highly substituted C–H bonds, but this tendency is counterbalanced by the steric demands of the catalyst,’ says Davies. The catalyst balances the steric and electronic effects, such that the carbene locked within the catalyst reacts preferentially with the C-2 carbon of pentane, creating a new C–C bond at that position.
‘What we have shown is that conceptually a catalyst can be designed to target specific C–H bonds of a substrate,’ Davies says. This opens the way to the design of new catalyst ligands of specific architecture to create scaffolds to target given C–H bonds.
Commenting on the work, Edward Anderson, who leads a synthetic organic chemistry group at the University of Oxford in the UK, says, ‘It’s extremely impressive not only to pick out this single site of reactivity from what appears to be many similar C–H bonds, but to do so with exquisite levels of enantio- and diastereoselectivity. Given the relative complexity of the products, the commonplace nature of alkanes as starting materials, and the range of functional groups tolerated in this chemistry, it’s likely to be of considerable use in stereoselective synthesis.’
References
K Liao et al, Nature, 2016, DOI: 10.1038/nature17651









No comments yet