Tool will help chemists pick the best solvent for their reaction
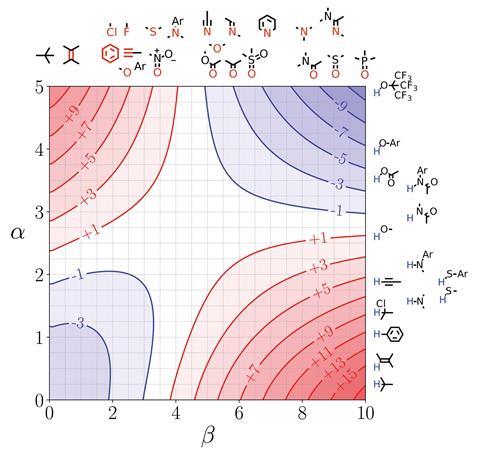
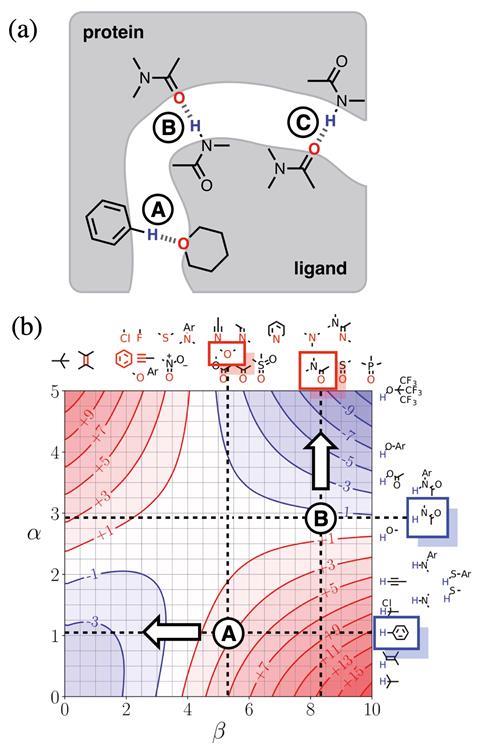
‘This is a step towards trying to encourage experimentalists to think about their problems based on theoretical concepts without having to engage with the full horror of theoretical calculations,’ says Christopher Hunter from the University of Cambridge, UK, about his team’s new model.1 Building on experimental studies of pairwise interactions between hydrogen bonded solutes,2 Hunter and his colleagues have developed a way to quantitatively predict how a solvent will affect non-covalent interactions and extended it to multiple interaction sites in any solvent environment. They also developed visual maps that direct experimentalists to which sites on a molecule they should tailor in terms of polarity to enhance desired interactions.
Non-covalent chemistry sits at the heart of most of the functional uses of organic molecules. Almost all of the interesting properties surrounding organic molecules emanate from how they interact with other molecules and since most applications take place in the solvent phase, understanding the effect of solvents on these interactions is useful to know.
For Hunter, the work is a labour of love. ‘I was trying to formulate how I could square the supramolecular chemistry literature, that was all in chloroform, with the biochemistry literature, all in water, in a model that treated both very different solvent environments in the same way and this was the result. It evolved from a qualitative model that was in a second-year undergraduate lecture course, becoming gradually more and more sophisticated to the point we’re at now.’
Referred to as ‘functional group interaction profiles’, the model unpicks individual points of interaction between molecules. ‘What are the properties of individual pieces of the molecule and how are they likely to affect the properties? My aim was to develop a rule of thumb approach for experimental organic chemists to use in the same way they use curly arrows to think about mechanism or conformational analysis to think about 3D structure. A chemist will have a model to think about those phenomena without resorting to a simulation.’
Design to dissolve
Hunter explains that his group has simplified interactions as a number of expressions. ‘Even though the underlying quantum mechanical calculations of the electrostatic surface and the profiles are complicated, the way all of that is subsumed into a fairly simple expression makes it very useable to non-experts. And the profiles themselves are very easy to interpret,’ comments chemical physics expert George Jackson of Imperial College London in the UK.
Jackson says chemists will be able to use it predictively because it gives information about the equilibrium constant and the binding constant between different molecular groups. ‘That’s extremely useful if you want to design something that binds tightly to a protein or if you want the interaction between the molecule and the solvent to be strong so it’s soluble. Things like being able to predict the solubility of drug molecules, the binding of molecules onto proteins, these are crucial bits of information that you need to know. There’s no point a drug being developed that binds effectively to a protein if it’s not going to dissolve in your stomach and go into your bloodstream.’
‘I think what an experimental chemist wants is a model that is going to tell them how their system is going to behave, really,’ adds Hunter. ‘We’re still a long way off theoretical methods being able to provide a reliable prediction on a complex system. I’ve tried to extract something that might be useful to an experimentalist from theoretical calculations. The proof of the pudding is whether people actually pick it up.’
References
1 M D Driver et al, Chem. Sci., 2020, 11, 4456 (DOI: 10.1039/d0sc01288b) (This article is open access.)
2 C A Hunter, Angew. Chem., Int. Ed., 2004, 43, 5310 (DOI: 10.1002/anie.200301739)











No comments yet