
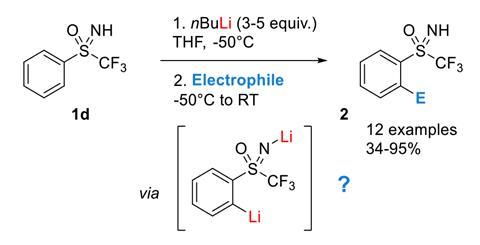
A number of surprisingly complex aggregates have been discovered in the directed ortho metalation, a classic reaction discovered more than 80 years ago. They are the reason this reaction requires a huge excess of butyllithium base: the aggregates ‘cannibalise’ the base and stop it from reacting.
Discovered independently by Georg Wittig and Henry Gilman in 1939, the directed ortho metalation is a type of electrophilic aromatic substitution. In it, an organolithium base such as n-butyllithium (BuLi) deprotonates an aromatic ring. This is directed by a heteroatom-containing substituent on the ring – such as a methoxy or an amide – so the reaction takes place exclusively ortho to this group. The aryllithium intermediate can then react with any number of electrophiles, making this an easy way to create functionalised aromatics.
‘Theoretically speaking, you just need one equivalent of base to remove one hydrogen,’ says Emmanuel Magnier from the University of Paris-Saclay in France. But common chemistry knowledge has it that this reaction needs at least two, sometimes up to five equivalents of BuLi – not a very atom-economic affair, Magnier points out. Organolithium compounds tend to create aggregates, but what exactly is consuming most of the base in this reaction has remained a mystery.
A team led by Magnier, Jacques Maddaluno and Hassan Oulyadi has now found various ‘base-eating’ aggregates in the ortho lithiation of an S-trifluoromethyl sulfoximine-substituted benzene.
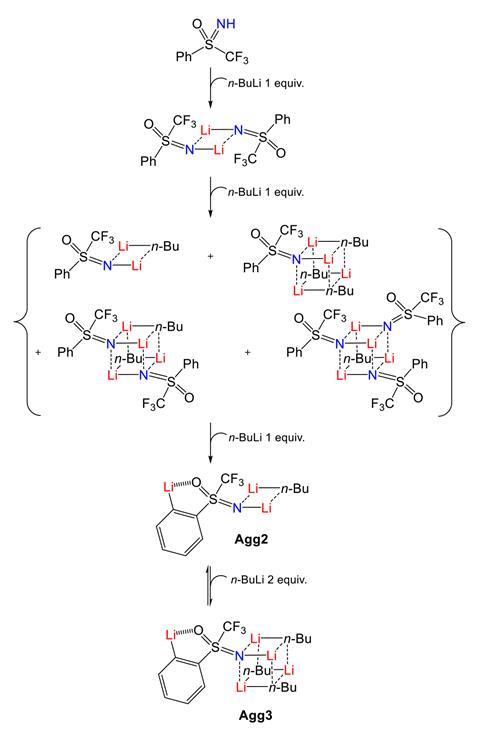
As expected, the first BuLi equivalent deprotonates the NH group. But as more BuLi is added, things get complicated. By combining sophisticated NMR experiments, isotopic labelling and density functional theory calculations, the researchers discovered three aggregates that incorporate up to three base molecules in various square and cubic arrangements. The base trapped in these compounds is completely unreactive, so the ortho deprotonation on the aromatic ring only happens when enough free BuLi becomes available.
And the aggregates have more surprises to offer: there are distinct homo- and hetero-chiral dimers with the chiral-at-sulfur substrate, and they protect the sulfur atom from nucleophilic attack making the reaction possible in the first place. For Magnier, the most unexpected thing was that the directing atom in the sulfoximine is in fact the oxygen. ‘I would have bet on the nitrogen,’ Magnier laughs.
Eva Hevia, an expert in organolithium chemistry at the University of Bern, Switzerland, is impressed by the ‘amazing level of detail’ the French team went into to uncover what consumes the base. ‘I think it’s fascinating on two fronts: first, because it helps us to understand why we actually need to use an excess [of BuLi] and how this excess is working. But secondly, because it shows the structural diversity and how much is still unknown about organolithium reagents that have been around for over 100 years.’
However, she points out, the results may not translate to other lithiations. ‘In organolithium chemistry, you change one variable – the solvent or the temperature, the concentration – and everything changes,’ Hevia says. Maddaluno agrees that the intermediates will be different depending on the substrate. Though as long as it has a substituent that can be deprotonated, aggregates are likely to form. ‘You can be almost sure that part of your base will be consumed by that,’ he says.
For the chemistry community, Hevia says, this raises the question: ‘What can we do to avoid the formation of these aggregates that cannibalise some of the reagent that you put in there?’ Maddaluno wants to see whether adding salts like lithium chloride could break up the aggregates. Magnier is excited to see whether running the reaction in a flow setup can suppress aggregate formation.
‘I see it as an inspiration for looking with much more detail into classical transformations that we take for granted,’ says Hevia.
References
M Hédouin et al, Angew. Chem., Int. Ed., 2022, DOI: 10.1002/anie.202214106














No comments yet