Neural network folds proteins a million times faster than its competitors
By  Katrina Krämer2019-05-08T09:30:00
Katrina Krämer2019-05-08T09:30:00
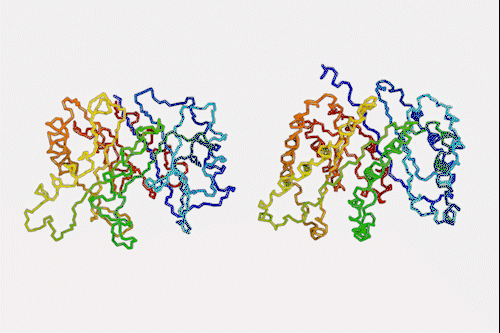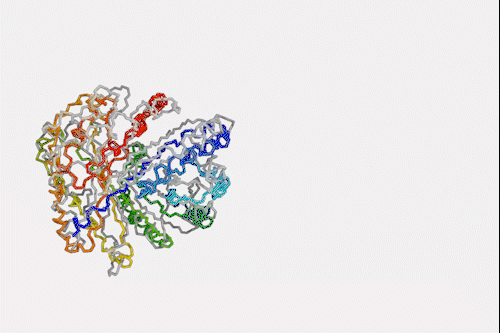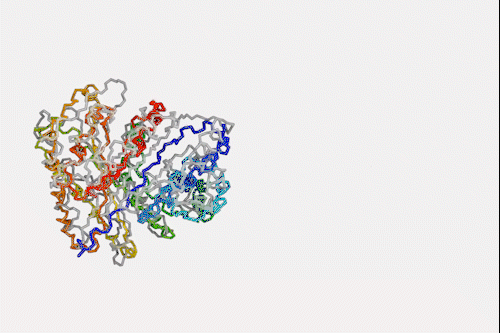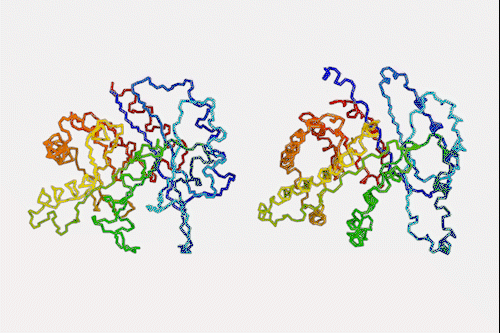
Machine learning algorithm that predicts protein structures in milliseconds could top next protein folding contest
Despite knowing next to nothing about chemistry or biology, a neural network can make a good stab at one of the toughest problems in biochemistry – predicting how a protein folds simply by looking at its amino acid sequence. The machine learning algorithm is a million times faster than other prediction programs, making it a hopeful to win next year’s worldwide protein folding championships.