Researchers in the US have produced remarkably realistic molecular dynamics simulations of how metal–organic frameworks (MOFs) adsorb water.1 ‘You can almost drink our water model because it’s so realistic,’ jokes Francesco Paesani, who led the study at the University of California, San Diego. ‘You should trust the simulations because they are really very, very close to what the reality is.’
Atmospheric water harvesting has emerged as a potential strategy for tackling fresh water shortages, particularly in areas far from the coast. ‘Having a material that can just capture water from the atmosphere, even at those lower humidity ranges, could be quite transformative,’ comments Paesani.
MOFs’ large surface area and wide range of tuneable properties make them appealing materials for atmospheric water harvesting. However, previous computational studies on water harvesting with MOFs often fail to realistically reflect experimental results. ‘The main challenges of simulations of water in MOFs is that water enters as a gas but then as it fills the pore, it starts developing liquid-like and even ice-like behaviour,’ explains Paesani.
Now, Paesani’s group has shown how performing classical molecular dynamics simulations using a potential energy function developed by the team called the MB-pol data-driven many-body potential, could address such challenges. The simulations report results much closer to experimental values than previous models and can ‘provide insights into what each water molecule does when it enters the pore,’ he says.
‘The MB-pol model developed by Paesani and co-workers provides outstanding agreement with experiment for a wide variety of properties of water,’ comments Randall Snurr, an expert in MOF modelling at Northwestern University in the US.
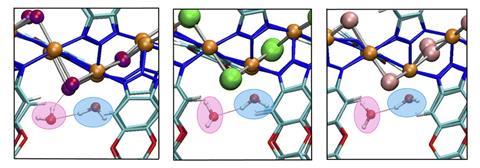
Paesani’s team tested the model on Ni2F2BTDD (X = F, Cl, Br), a promising halide-exchanged MOF2 that ‘shows the best performance in the range of relative humidity between 20–30%,’ says Paesani. ‘It is a MOF that is able to adsorb a large amount of water molecules relative to the mass of the MOF within that humidity range.’
They found that exchanging the halide atoms in Ni2X2BTDD resulted in three MOFs with different properties and water adsorption capabilities. While Ni2F2BTDD and Ni2Cl2BTDD adsorb over 100% of their weight in water at 32% relative humidity, Ni2Br2BTDD adsorbs 64% of its weight in water below 25% relative humidity; one of the best water uptake values ever reported.
What is particularly interesting, says Snurr, is the study found that ‘some properties, such as the enthalpy of adsorption, do not follow the trend of F, Cl, Br’. ‘From the molecular-level modelling reported here, the [researchers] are able to provide useful insights into these unexpected trends.’
Paesani is confident their model will influence future research on atmospheric water harvesting materials. ‘Experimentalists can use our data and be sure that what we see is exactly reflecting what they measure in experiment,’ he explains. This should ‘help speed up the development of materials without going through the synthetic process, which can be time consuming and much more expensive.’
References
1 K M Huntera and F Paesani, Chem. Sci., 2024, DOI: 10.1039/d3sc06162k
2 J J Oppenheim et al, J. Am. Chem. Soc., 2021, 143, 16343 (DOI: 10.1021/jacs.1c07449)











No comments yet