Streamlined method of constructing complex molecules could help tap nature's bounty.
A radically different approach to constructing complex molecules could help to tap the pharmaceutical potential of natural products. The concept, devised by Phil Baran and colleagues at Scripps Institute, La Jolla, California, promises to generate natural products in much larger amounts than conventional methods, making biological testing much easier for drug discovery scientists.
Most total syntheses, which assemble complicated carbon-based molecules from relatively simple building blocks, make liberal use of protecting groups. These chemical shields prevent completed parts of a molecule being altered while chemists are still tinkering with other sections that are still under construction. However, adding and removing protecting groups can add many steps to a synthesis, cutting overall yields drastically.
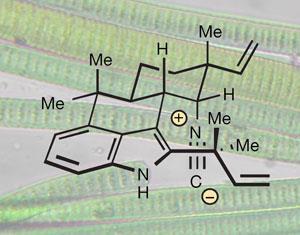
Baran’s team have now made a collection of marine natural products without using a single protecting group. Instead, they take advantage of the intrinsic reactivity of the molecule’s different functional groups.
This unconventional approach delivered several grams of compounds such as ambiguine and welwitindolinone in less than 10 steps. What’s more, the reactions were enatioselective - they made only the preferred mirror-image form, or enantiomer, of the molecule, instead of a racemic mixture containing equal amounts of both enantiomers. This is a vast improvement over traditional syntheses, which make milligrams of racemic product in around 30 steps.
Baran’s strategy is summed up by eight guidelines.1 These include maximising the percentage of carbon-carbon bond forming reactions; using cascade reactions that form several bonds at once; cutting reduction or oxidation reactions to a minimum; and avoiding protecting group chemistry. ’You could say we are re-choreographing natural product synthesis,’ Baran told Chemistry World.
The approach has been successful for other syntheses. ’Just recently, we have made a natural product with ten nitrogen atoms in without using protecting group chemistry, which is an amazing feat,’ said Baran.
The Scripps team are confident that this way of thinking will be widely used in natural product synthesis. ’We are taught that the way to solve these problems is to protect functionality rather than embrace it,’ Baran said. ’But I think the day will come when chemists can synthesise Taxol without using protecting group techniques.’
Reinhard Hoffmann, at the University of Marburg, Germany, said the strategy was a move in the right direction. ’As more examples with the stringency of Baran’s work are known, it will induce others to follow and chemists will no longer get away with doing syntheses in which protecting groups and redox steps abound.’
Keeping the oxidation level of intermediates the same, or allowing it to increase step by step throughout the synthesis, is crucial, added Hoffmann. ’Baran’s method makes us think about what steps we can avoid in a synthesis, leaving only the obligatory steps - those which form the skeleton of the product,’ said Hoffmann.
Baran is not advocating that protecting group chemistry will become redundant. ’For the production of some compounds, like polyketides and polypeptides, the use of protecting groups will still be the most efficient route,’ he said.
’But there is this far-ranging and damaging perception that natural products are too complex to be used in a drug discovery setting despite their overwhelming track record. I think if our work has helped in even a small way to revive their use, then we have served our purpose.’
Alison Stoddart
References
Nature446, 404






No comments yet