
Calculation will help users recognise when they need to retrain a neural network
Researchers in the US have developed a better way to measure the confidence of neural networks’ predictions. ‘When we want to go out and discover new materials, one of the biggest challenges is how do we know if we trust the model or not,’ says Heather Kulik from the Massachusetts Institute of Technology, who led the research. ‘This is something that extends across challenges in chemical discovery, which was the focus of our work, to other areas, such as image recognition or other applications of machine learning.’
Neural network models emulate how the brain learns and processes data. A web of virtual neurons responds to different features of the input, for example a molecule’s connectivity, size and charge. These neurons are usually organised in layers, with the output of one layer providing a more refined input for the next, until the output of the final layer is processed into a result, such as a chemical property. The network is trained on a set of known examples by modifying the responses of its neurons until it gives the correct answers, after which it’s expected to make good predictions for new data.
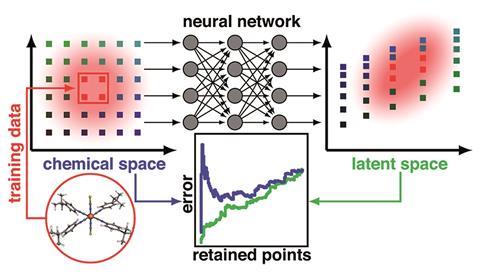
The new research by Kulik and her team looked at how the confidence of those predictions is measured. One way is to measure variations in a prediction between very similar models, for example in an ensemble that was trained together, or one network with connections randomly dropped out. ‘When models disagree that means that none of the models really know the answer,’ explains Kulik. Alternatively, it can be supposed that a model won’t work well with an input that’s very different from the data it was trained on, which can be expressed as a distance in a feature space (ie the difference between two points in a molecular representation). Unlike other methods, calculating this is cheap as it doesn’t involve re-running or retraining the model. However, it only works well if the features describing the input are strongly related to the properties being predicted. Similarities and differences in irrelevant features could make a molecule seem closer or further from the training data than it really is.
Kulik’s group realised that the neural network accounts for this, encoding a more relevant description of the molecule in the last layer of neurons before the output, known as the latent space. ‘What the neural net is essentially doing is stripping away details from the features that are not helpful and amplifying the most important ones, and by the time we get to the latent space, it’s basically the way the model sees the molecule to make the prediction,’ Kulik explains. The distance in this space can then act as a better measure of how different a given bit of input is from the training data. ‘We got talking about how points move around both during training and as you pass through the layers of the model, and that it might be a higher fidelity or better way of understanding what the model knows about a new molecule. It was very intuitive once we landed on it.’
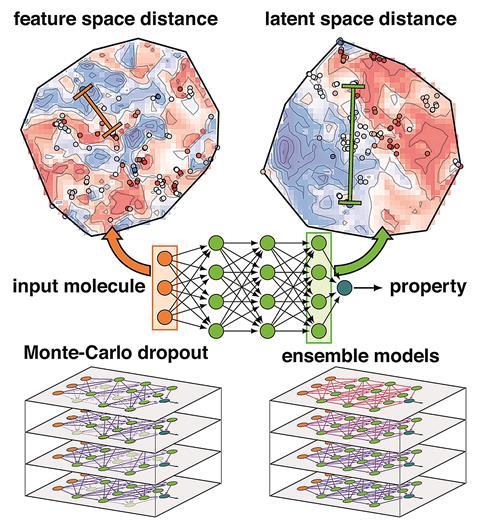
The latent space distance gave more accurate estimates of the uncertainty than the ensemble, drop-out and feature-space techniques in two quite different problems: predicting spin-splitting in inorganic complexes, and the atomisation energies of organic molecules. It even gave useful results when tested for an image recognition problem. ‘It [the latent space distance] is a basic property of neural networks that does not depend on the underlying application,’ notes Clémence Corminboeuf, who develops theoretical chemistry techniques at the Swiss Federal Institute of Technology in Lausanne (EPFL). ‘Not all the machine learning models used in chemistry are based on neural networks. Yet, this approach will inspire the use of similar metrics.’
A new routine
Understanding where a model is confident can help refine it. For example, it can be retrained only in areas where it performs well, giving a specialised but reliable tool. Alternatively, retraining can focus on areas where the model is weaker, making it more general. The latent space measure gave better results in both cases, and its efficiency, particularly compared to ensemble methods, is an advantage in automated active learning. ‘You want a fairly tight loop between finding the uncertain points, training the model, and then repeating that process very iteratively,’ says Kulik.
The importance of measuring chemistry models’ confidence may not yet be widely appreciated. ‘If you take a look at the literature, it’s not uncommon that people make predictions with machine learning models or use them without ever really knowing how well a model will work, and how well a model will generalise,’ says Kulik. Corminboeuf says something similar: ‘Evaluating the errors associated with predictions on new molecules or conformations is clearly not yet a routine procedure, but uncertainty metrics are essential to evaluate the reliability of these predictions. They will and should be used more and more.’














No comments yet