Agelastatins A-F
Agelastatins A-F
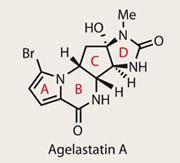
After a target has been synthesised, and the question of ’can we make this?’ has been answered, perhaps the most important remaining question is ’how did nature make it?’ Sometimes the biosynthetic route is obvious, but nature often works in mysterious ways, so no room should be left for supposition. In the case of the agelastatin family, syntheses of all six variants have followed thick and fast since their isolation, which started with agelastatin A in 1993.1,2 This is no doubt down to their intriguing biological activity, which includes ’significant antitumor activity’, as well as inhibiting glycogen synthase kinase-3b . The latter might not ring many bells, but it’s implicated in Alzheimer’s disease and bipolar disorder, making it an interesting biological target.
But the question of the agelostatins’ biosynthetic origin has yet to be answered. Therefore, a team led by Mohammad Movassaghi at the Massachusetts Institute of Technology, US, decided to attempt a chemical synthesis that follows the suspected biosynthesis, thereby testing the proposal and hopefully enabling a succinct synthesis.3
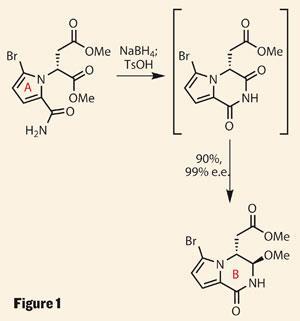
They kicked off with a pyrrole ring fragment derived from d-aspartic acid, which was selectively brominated and amidated. The resulting tricarbonyl compound looks primed for all sorts of chemistry, but the intended cyclisation-reduction sequence gave a cracking yield and selectivity (figure 1). Importantly, Movassaghi describes this chemistry on multigram scale - key for the team’s divergent approach to the whole family of natural products.
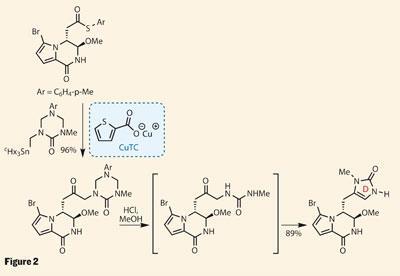
An unlikely couple
With two rings in place, the group pressed on, planning to append the D ring next (figure 2). Their aim was to use the remaining methyl ester as a handle to add a triazone - a six-membered ring containing three nitrogen atoms and a carbonyl group - but the coupling chemistry was recalcitrant to say the least. Their response was to replace the methyl ester with a thioester analogue. This coupled readily with an alkyl stannane, using stoichiometric amounts of the slightly expensive (but amazingly useful) copper thiophenecarboxylate (CuTC) as the mediator rather than a more typical palladium catalyst.
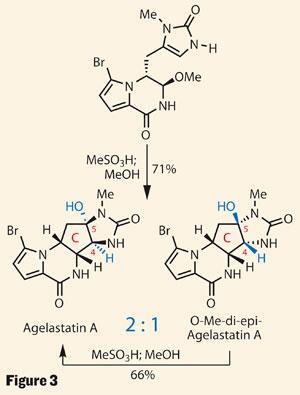
On first inspection, it’s hard to see why the group opted to attach the triazone, as this ring doesn’t feature in the target. However, a little acid unravelled this exotic heterocycle, leaving behind a more familiar urea. This then cyclised back on itself, neatly forming the imidazolone D ring. More importantly, this intermediate also features in the proposed biosynthesis, giving Movassaghi the perfect platform to test his biomimetic plans.
All that remained was to form the highly substituted cyclopentane C ring which, with its four contiguous stereocentres, had formed the crux of many prior syntheses. In contrast, the team hoped that by aping nature, these stereocentres would simply fall into place.
A nip of organic acid in methanol was enough to achieve a very respectable yield of agelastatin A, along with a diastereoisomer in which the newly formed bridgehead stereocentres (C-4 and C-5) are inverted, lending much credence to the team’s biosynthetic proposals. Crucially, the group again performed this chemistry on gram scale, allowing them to make agelastatins B, C and E from the same intermediate, as well as D and F by a related route.
I haven’t got the space here to go into the details of their biosynthetic investigation - that’s what a six page full paper is for - but I admire their approach hugely.
Paul Docherty is a science writer and blogger based in London, UK
References
1 M D'Ambrosio et al, J. Chem. Soc., Chem. Commun., 1993, 1305.
2 M D'Ambrosio et al, Helv. Chim. Acta, 1996, 79, 727.
3 M Movassaghi, D S Siegel, S Han, Chem. Sci., 2010, DOI: 10.1039/c0sc00351d












No comments yet