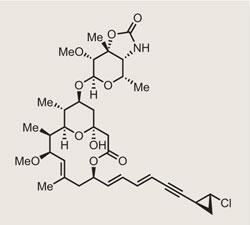
Callipeltoside A
Harvard University’s David Evans is renowned for his prowess with an aldol reaction, and he has used the endlessly flexible transformation to make a diverse range of natural products.
His latest target, callipeltoside A,1 screams ’aldol me up’ more than most compounds, and it has attracted the attention of all the usual suspects since its isolation in 1996.2,3,4
However, like watching a seasoned sportsman warming up for what promises to be a classic game, there’s a real feeling of anticipation about how he’ll apply the reaction to this challenge.
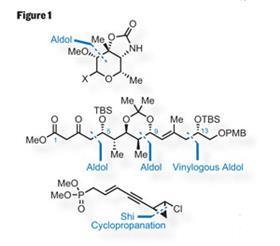
The more obvious disconnections (figure 1) remove the sugar unit - inventively called callipeltose - from the top of the molecule. Cutting the crazy-looking chloro-cyclopropane from the bottom leaves a macrocyclic lactone
(or macrolactone) in the middle. Opening up this ring leaves a linear structure, with hydroxyl and methyl groups sticking up and down all over the place - ideal for an aldol or three.
Slowly does it
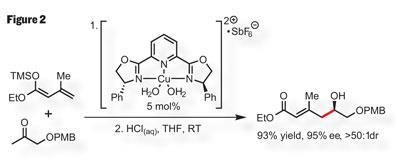
So, to the first aldol - a catalytic, asymmetric, vinylogous aldol to be precise. Evans uses two non-chiral starting materials, coupling them using a C2 -symmetric ’copper-box’ catalyst (figure 2). Evans has pioneered this reaction over the years,5 and in this case it provides a tricky stereocentre in high yield, along with good control of asymmetry and geometry in the remaining olefin. What more can be asked? The only catch is that he needs to add both starting materials quite slowly. Definitely one of those days in the lab when you hope the syringe pump is free, or that you’ve got a good book to read.
Aldol number two is in the synthesis of the callipeltose sugar unit. As many an organic chemist knows, sugars can both be your friends and mortal enemies - fantastic as sources for chirality, but a pain in the proverbial to synthesise. However, there are plenty of precedents for using aldol chemistry to produce these targets, particularly from Evans’ prot?g? David MacMillan,6 now at Princeton.
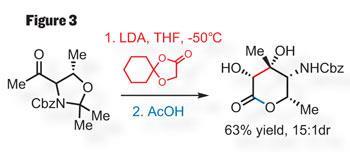
In this case, the stereochemistry of the aldol reaction is controlled by the ketone substrate (figure 3). Adding the lithium enolate of the ester partner prompts an efficient aldol from a cyclic transitions state, giving the aldol adduct in good yield with excellent stereochemical control. Removing both ketal-type protecting groups with acetic acid then triggers a lactonisation to give the six-membered lactone analogue of callipeltose.
However, it took five steps to make the starting material for this aldol, adding up to twelve linear steps to produce the substrate required for coupling. I don’t think that this shows an inadequacy in the design of the synthesis - rather, it highlights the fact that we still find certain structural motifs a real challenge.
Crazy rings
I’ll leave the rest of the aldols for now, and turn to an asymmetric cyclopropanation. The first time I clapped eyes on callipeltoside A, I was flabbergasted by the crazy chlorocyclopropane subunit. I wondered if simply looking at it the wrong way would be enough to make it fall apart. But it seems that this is one of those (all too common) situations where nature makes me feel like an idiot.
So how does one make an enantiomerically-enriched chlorocyclopropane? Evans tried several possibilities based on the Simmons-Smith reaction, which adds a carbene - generated from diiodomethane and diethyl zinc - to an unsaturated carbon-carbon bond. He finally struck gold with chemistry developed by Yian Shi at Colorado State University,7 who found that adding an equivalent of trifluoroacetic acid to the reaction could improve yields - in this particular case, from a measly 10 per cent to a rather satisfying 82 per cent.
Overall, this is a fantastic blend of Evans’ own chemistry, where applicable, and that of others when more appropriate. That balance alone makes this a superb synthesis.
Paul Docherty is a DPhil student at the University of Oxford, UK
References
1 D A Evans et al,Tetrahedron, 2008, 64, 4671
2 B M Trost et al,Angew. Chem. Int. Ed., 2002, 41, 841
3 I Paterson et al,Org. Lett., 2003, 5, 4477
4 J S Panek et al,Org. Lett., 2004, 6, 4383
5 D A Evans et al,J. Am. Chem. Soc., 1999, 121, 669
6 D W C MacMillan et al,Science, 2004, 305, 1752
7 Y Shi et al,Tetrahedron Lett., 1998, 39, 8621










No comments yet