Cochleamycin A
Cochleamycin A
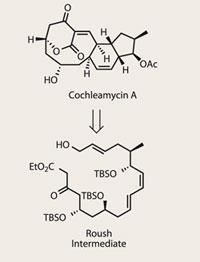
When it comes to making large natural products, different researchers will often propose identical ’end-game’ strategies to complete the target. This similarity isn’t too surprising, as there is often a stand-out feature that looks amenable to synthesis, such as a lactone or an olefin. This fact leads to many ’formal’ syntheses of large natural products - in which the target is not the natural product, but a late-stage intermediate in an existing piece of work.
In the case of cochleamycin A, its potent biological profile has attracted numerous efforts in the past few years, with several total syntheses published. Before we get into the chemistry, it’s worth examining the biological profile - in this case significant leukaemia-cell bashing, along with that perennial grant-winner, antibiotic activity.1 One synthesis in particular, by Scripps-based William Roush,2 caught the eye of Daesung Lee at the University of Illinois, Chicago, who decided to target an advanced intermediate in Roush’s route.3
Juggling act
As is usual for a target like cochleamycin A, the initial work was synthesis of smaller constituent fragments. The necessary asymmetry was introduced using a reaction that I wasn’t too familiar with - adding the three carbon allyl unit to an aldehyde using a pseudoephedrine-derived strained silacycle. Although the yield and enantiomeric excess were only moderate, Lee explains that other, more popular methods (such as the Keck or Roush allylations) were even less effective. Key to this reaction is the use of an organo-silicon reagent, exemplifying the differences in reactivity between carbon and its less popular row three neighbour.
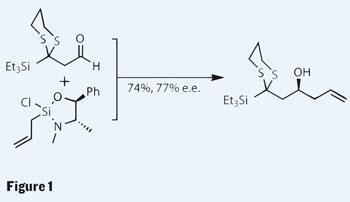
Lee seems quite the expert with a silicon atom, and shows a little more of his expertise with it as he juggles the triethylsilane group onto the hydroxyl via a Brook rearrangement, forming an alkyl lithium intermediate with which they were able to further functionalise the molecule. The other main component was produced in five steps, also using an asymmetric allylation reaction to introduce the necessary stereochemistry. This brings us neatly to the unification of the two halves, using a tethered metathesis protocol, which is going to take a bit of explaining.
Firstly, the group roughly stitched the two components together using an easily cleaved silicon group. This was done using a rather exotic-looking trialkynyl cyclopentyl silane reagent, rather than the more familiar halo-silanes found in every reagent fridge (often brown, syrupy and with floaty-bits...). Reacting this silane with a suitable alcohol, treated with base to form an alkoxide, results in an addition reaction to give a hypervalent silicon intermediate. This kicks out an acetylenide anion, which deprotonates a further molecule of alcohol, forming the next attacking alkoxide. The acetylene rejected is presumably lost as a gas, which coupled with the need for only catalytic base provides a rather neat reaction. I hadn’t seen this type of reagent used before (and I used a lot of silyl groups in my DPhil), so I wasn’t too surprised to see that it was recently developed by Lee himself.4
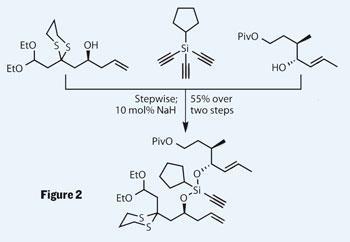
The group use this protocol to temporarily tie the two complex alcohols together in a stepwise process, leaving one acetylene moiety in place. This was key, as it participated in a very selective metathesis reaction, providing a diene product with excellent control of geometry, favouring the E,Z -configuration required. This reaction works in several steps, the first two involving Grubbs catalysed metathesis reactions as first one olefin and then the other reacts with the central alkyne to build the desired diene. (Of course, this kind of control doesn’t happen by accident - the group has been working on this kind of chemistry for some time.5) A little TBAF (tetra-butyl ammonium fluoride) did its usual trick and removed the silicon tether in the same manner as the protecting groups it more frequently knobbles.
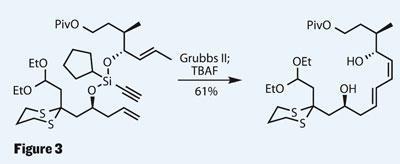
Although this synthesis doesn’t complete the natural product, it’s more than fair for the group to accept Roush’s route as having the ideal end-game. What it does do is exemplify both modern and traditional uses of organo-silicon chemistry in organic synthesis.
Paul Docherty is a medicinal chemist based in London, UK
References
1 K Shindo, M Matsuoka and H Kawai, J. Antibiot., 1996, 49, 241
2 T A Dineen and W R Roush, Org. Lett., 2004, 6, 2043 (DOI: 10.1021/ol049331x)
3 S Mukherjee and D Lee, Org. Lett., 2009, DOI: 10.1021/ol900923c
4 J B Grimm and D Lee, J. Org. Chem., 2004, 69, 8967 (DOI: 10.1021/jo048908l)
5 S V Maifeld and D Lee, Chem. Eur. J., 2005, 11, 6118 (DOI: 10.1002/chem.200500407)












No comments yet