Haplophytine
Haplophytine
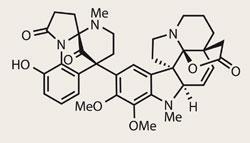
It’s been a while since I’ve seen such a battle for the ’first publication’ of a molecule as has recently been witnessed for haplophytine. This deca-cyclic (not a term I get to use enough) target has been exceptionally stubborn, with its synthesis campaign running into a quarter century. However, like British buses, you wait 25 years, and then two arrive together.
It was Tohru Fukuyama of Tohoku University in Japan who claimed the ribbon for first-to-publish;1 but at only four-months difference, K C Nicolaou and his team (co-led by David Chen) at the A*STAR institute in Singapore can’t feel particularly left behind.
The A*STAR team published their progress towards the molecule in 2008, in the form of aspidophytine - a distinct natural product which makes up half of haplophytine.2 However, that leaves an equally challenging half and the coupling of the fragments for this most recent paper.3
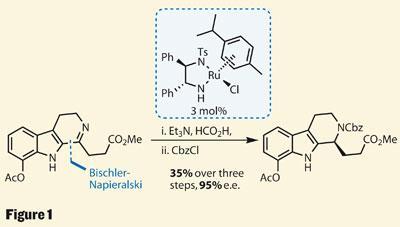
Synthesis of the left hand side (LHS) requires the creation of four rings, so the group made life as easy as possible by starting with two in the starting materials. A third ring was soon added to this indole fragment by imine formation; a quick start, but lacking the required asymmetry. However, the freshly installed imine functionality was very susceptible to asymmetric reduction - a perfect system for Ryoji Noyori’s Nobel prize winning ruthenium chemistry, which delivered the asymmetry in excellent enantiomeric excess (figure 1).
Great minds
Like Fukuyama, the team chose this point to install a second, catechol-type indole system. Nicolaou used the oxidative properties of hypervalent iodine (in this case, phenyliodine-bis-trifluoroacetate, or PIFA) to add the catechol.
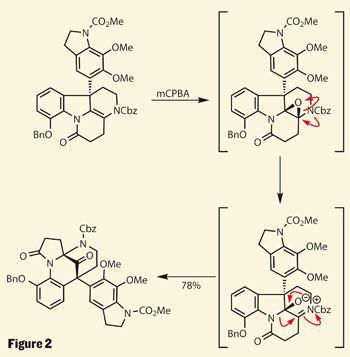
The intermediate in their flask at this stage was almost identical to one found in Fukuyama’s lab earlier this year - and their route to install a sensitive ketone is similar (figure 2). Both rely upon a propensity for the natural product to rearrange (via a 1,2-shift), allowing the chemists to target the synthetically less-challenging structure, and then trigger the rearrangement. Both groups even used the same reagent to form a fragile intermediate epoxide, activating the expansion of one ring and contraction of another, and completing the LHS.
Never ever
Rather than coupling the two halves together in the latter stages of the synthesis, Nicolaou chose to construct the RHS around the catechol-indole (figure 3). This strategy firstly involved a very complex Suzuki coupling (requiring three equivalents of thallium ethoxide - a regent on my ’never, ever’ list), followed by an intriguing ring formation. A little triflic anhydride (on the ’steady-on-now’ list) promoted this second reaction, and again left an imine.
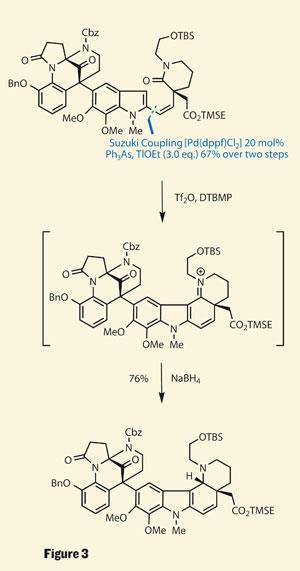
Again, their plan was a diastereoselective reduction, but this time the conformation of the molecule (substrate control for those into jargon) allowed use of simple sodium borohydride to impart the desired stereocentre. It’s great when nature helps out. This approach was also key to their next ring-construction, using free radical chemistry to form the second-to-last ring.
So what about the last ring? Again, twenty-five years of research allowed the group some insight into possible strategies, and like Fukuyama, they used an approach developed by Harvard’s E J Corey.4 And this exemplifies the tactic used by both successful groups - sharing methods to develop a route containing the best ideas available. The fact that the routes are so similar in places is simply a reflection that these are the ideas that work.
Paul Docherty is a medicinal chemist based in London, UK
References
1 H Ueda et al, Angew. Chem. Int. Ed., 2009, DOI: 10.1002/ange.200902192
2 K C Nicolaou, S Dalby and U Majumder, J. Am. Chem. Soc., 2008, 130, 14942 (DOI: 10.1021/ja806176w)
3 K C Nicolaou et al, Angew. Chem. Int. Ed., 2009, DOI: 10.1002/anie.200904588
4 F He et al, J. Am. Chem. Soc., 1999, 121, 6771 (DOI: 10.1021/ja9915201)












No comments yet