Maduropeptin chromophore
Maduropeptin chromophore
This month’s target couldn’t be much more different from last month’s compact alkaloid; although the gross number of stereocenters isn’t vastly removed, the class of natural product certainly is. The maduropeptin chromophore (the active component of a chromopore-protein complex, noted as for its potent antitumor and antibiotic activity) is built of two distinct domains - a relatively ’synthesis-friendly’ glycoside chain appended to an altogether more harrowing core featuring a nine-membered diyne and a 15-membered macrolactam. This first successful approach to the molecule, led by Itaru Sato and Masahiro Hirama at Tohoku University, Japan, not only completed an heroic synthesis, but also corrected the stereochemistry of the sugar moiety, as they report in a couple of recent papers.1,2

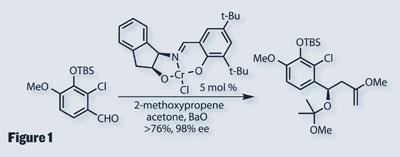
Synthesis started at the core, with a rather complex tri-substituted benzaldehyde, using chemistry developed by Harvard’s Eric Jacobsen, introducing asymmetry as well as installing a synthetically useful enol-ether (figure 1). To do this, the group used a chiral, tridentate chromium(iii) catalyst to perform an asymmetric ene reaction in high yield and enantiomeric excess.3 Importantly, this reaction was quite happy to scale-up to the big flasks, allowing the group to move on to the diyne section.
Double bond dilemma
Building the diyne - by coupling a series of pre-formed fragments - was completed over a rather lengthy campaign; not because the route was inefficient, but because such structures are frequently unstable. Indeed, inadvertently introducing an alkene between C4 and C5 is all that is necessary to trigger an intramolecular cycloaddition - something that the team were at all costs to avoid.
As an interesting aside, it’s this proclivity for cycloaddition that is behind the molecule’s intriguing biological activity. A loss of methanol allows the compound to form a rather strained aziridine intermediate, which then does the afore-mentioned cycloaddition (figure 2). As there is an excess of electrons, the initial product is a ’p -benzyne biradical’, an extremely reactive intermediate with a voracious appetite for hydrogen atoms, which it feeds by cleaving double-stranded DNA.
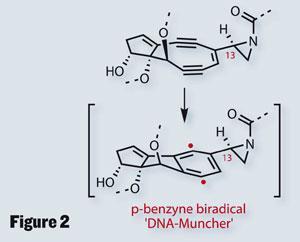
As interesting as this is, the group was keen to prevent such chemistry - the problem being their need to install an alkene across the neighbouring C4-C13 bond. Key to such a strategy is control, especially as they planned an elimination-type approach. Common, base-induced chemistry would bring me out in a nervous sweat in this situation, so I was intrigued by their approach - a samarium-diiodide promoted 1,2-elimination of two adjacent benzoates (figure 3).
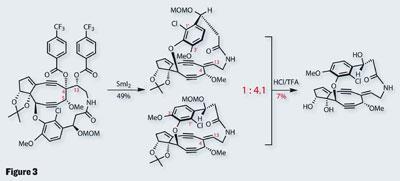
This methodology was developed in-house, but has yet to see publication, something I’m eager to read as the selectivity is pretty good, with a 49 per cent yield. However, a complication emerged at this point, as the product is isolated as a mixture of two atropisomers, formed due to the restricted rotation of the aromatic ring. Their brief joy at being able to separate these two must have turned to dismay as they discovered that interconversion at room temperature was unpreventable. However, the situation was rescued when it was found that global deprotection of the mixture gave only the desired product, although unfortunately in a fairly appalling yield.
In the more recent paper, the group described a solution to that distressing yield, using a stepwise deprotection protocol. Coupling the core with the glycoside moiety suggested by the isolation assignment was rather successful in terms of efficiency (using the Schmidt protocol, for the sugar-bashers out there), but less so once the protecting groups were removed, as the NMR of the synthetic and natural material didn’t match. The inconsistency was most obvious in those protons found on the sugar, so the group needed to reassign that moiety. Switching the sugar for its enantiomer, the coupling reaction wasn’t quite as efficient as before, but with a matching NMR, I guess the group didn’t mind, as the result after deprotection was the now reassigned target, and completion of a very volatile molecule.
Paul Docherty is a medicinal chemist based in London, UK
References
J. Am. Chem. Soc., 14184 (DOI: 10.1021/ja076671f) et al 124, 2882 (DOI: 10.1021/ja025588j)












No comments yet