Paul Docherty marvels at a parade of intricate radical reactions
Time and time again, traditional Chinese medicine turns up new, biologically active natural products. Often the origin and efficacy are already suggested, speeding up the search for active ingredients and narrowing the list of components to isolate.
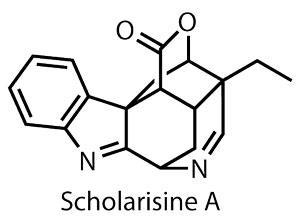
Scholarisine?A is one such species, identified from a traditional treatment for respiratory ailments in 2008, but not as yet individually analysed for biological efficacy.1 However, the synthetic challenge is already evident, with a complex cage structure bearing several tertiary and quaternary centres.
This isn’t entirely new territory – a team led by Amos Smith at the University of Pennsylvania, US, completed a neat but lengthy synthesis of scholarisine A last year.2 But at 25 steps, there is certainly room for Scott Synder and Myles Smith at Columbia University, New York, to improve on it.3
The team produces an enone dienophile bearing a chiral oxazolidine ring derived from the amino acid d-serine. Using the ever-popular Diels–Alder cycloaddition (figure 1) and a somewhat unusual cyclic diene (methyl coumalate), they build a neat bicyclic intermediate, bearing an internal olefin and an exocyclic carbonyl in useful locations. The oxazolidine controls the reaction geometry, before being broken open with a little acid, and the revealed terminal hydroxyl displaced with bromine.
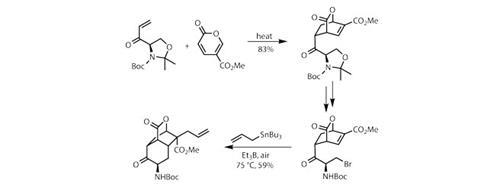
The stage is then set for the first of a trio of free radical reactions – chemistry involving short-lived intermediates with unpaired electrons. To generate the radicals, the team uses triethylborane under an air atmosphere (figure 1). The borane reacts with dioxygen to make a radical, which then rips off the bromine atom, making an alkyl radical that immediately closes down onto the bicyclic enone to form another six-membered ring. The resulting radical intermediate reacts with an allyl stannane to fix an allyl group to the tricyclic core. This somewhat blunt approach allows the team to construct a highly congested quaternary centre in excellent stereoselectivity and yield.
After this swift progress, the team faces a thorny problem. The stereochemistry of the primary amine needs to be inverted. The more obvious routes to this goal (deprotonating the a-proton) are foiled by competing side reactions. However, the amine turns out to be prone to radical oxidation, forming an amino-enone. The team capitalises on this feature by oxidising the amine with tetramethyl guanidine (TMG) and the stable radical oxidant TEMPO (2,2,6,6-tetramethylpiperidine-N-oxyl). The team exposes the amino-enone product to a deprotection–reduction–cyclisation sequence, which generates a six-membered lactam ring, completing the key cage structure of scholarisine A.
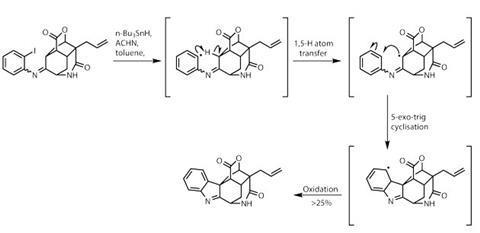
Rings a-plenty, but there are several more to deliver. Condensing the ketone with 2-iodoaniline adds the required aryl ring, setting the stage for a second radical cascade (figure?2). The iodine provides an easy site to form an aryl radical, which abstracts a hydrogen from the nearby cage system. That alkyl radical then attacks back into the aryl ring, before being oxidised to form the indole group found in the target. The overall yield for this unconventional process is low, but it is operationally extremely efficient, as so few transformations generate significant complexity.
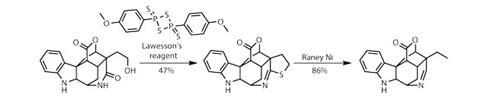
With the target still several steps away, treating the remaining terminal olefin with ozone under reducing conditions delivers a terminal hydroxyl, but also reduces the indole to an indoline. Using the alcohol as a handle, the team directs the fearsome-smelling Lawesson’s reagent to cyclise onto the lactam carbonyl, producing a tetrahydrothiophene ring (figure 3). This might not seem very productive, but cleaving out the sulfur atom with Raney nickel reveals the ethyl side chain and imine functionality found in the target. Finally, a little oxidation with iodosobenzene restores the indole, completing a breath-taking synthesis of scholarisine A in only 14 steps.
Paul Docherty is a science writer based in Leiden, the Netherlands












No comments yet