Cage-structured natural products are some of the most appealing targets for organic chemists
Cage-structured natural products are some of the most appealing (if perhaps not appetising) targets for organic chemists – perhaps due to their obvious intricacy of form, but also because of their structural rigidity. This inflexibility can be carried through the intermediates leading to their synthesis, posing an interesting challenge to the synthetic chemist. A recent synthesis of
vincorine, by Dawei Ma and his team at the Shanghai Institute of Organic Chemistry in China, posits an interesting strategy to these cages. By forming as much of the cage in one transformation as possible, the risk of encountering an intractable intermediate is reduced.
1The team began its synthetic odyssey with a rather neat piece of palladium catalysis – an alkenylation performed on a relatively simple and commercially available tryptamine. At the scale of the second step in a sequence (following Boc protection of the tryptamine), using rare metals can be financially challenging – even in catalytic quantities. Thankfully the chemistry was successful using simple palladium(II) acetate.2
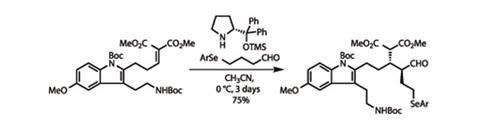
This approach may still have cost a little, but it saved a lot of time. The team required only four more steps to the get to the next key coupling reaction. Having added an a,ß-unsaturated dimethylmalonate group, they used a common proline-derived organocatalyst to engender enantio- and diastereoselectivity in the addition of an enamine derived from a relatively aldehyde, creating two new stereocentres in the process (figure 1).3
Keener readers will have noticed that the aldehyde sidechain has a rather unusual terminus – an aryl selenyl group. Like its group 16 partner sulfur, selenium(II) forms relatively stable bonds to hydrocarbon groups. However, higher oxidation states are less stable, and make for exceptionally good leaving groups. Adding a little
mCPBA was enough to oxidise the aryl selenide up to its Se(IV) oxidation state. It could then be removed entirely by working up the reaction with a little base. As expected, the elimination generated a new olefin group, but unfortunately only in a 1.7:1 E:Z ratio. However, the team managed to improve this to a far more satisfactory 30:1 by exposing the mixture to the correct wavelength of UV radiation to isomerise the new double bond.
A couple more steps took the team to the key step in their synthesis – a remarkable domino reaction sequence building two rings in one process. Treating the substrate with a strong base deprotonated the malonate group. This prompted the team’s showcase oxidative coupling reaction with the indole ring, forming an intermediate imine. The pendant amine group then closed down on this imine, neatly completing a second ring in one pot (figure 2).
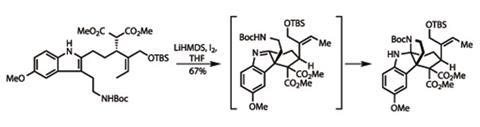
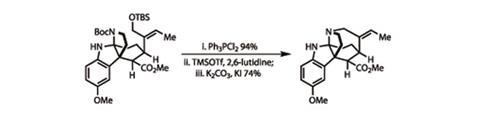
As I’ve mentioned in some of my previous columns, this is often the point where otherwise neat syntheses go astray, as the product of the key step requires significant elaboration to get to the target. Often these elaborations look small (one carbon homologations are common), but the steps add up. Ma’s synthesis doesn’t fall into that trap, though – taking only five more steps to get to vincorine. Of these, I particularly liked the final ring closure, completing the core cage structure. Working from a pair of protected heteroatoms, the team first used triphenylphosphine dichloride to remove the silyl protecting group from the remaining hydroxyl group, and transform this into a primary alkyl chloride in one high-yielding step.
4 The alkyl chloride was then further activated using potassium iodide through an in situ Finkelstein reaction, in which the chloride group is substituted for a more reactive iodide group, enhancing the ring formation step (figure 3).
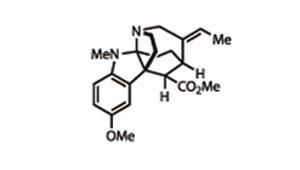
The team’s final task was to add one last methyl group to the aniline nitrogen, which they did by reductive amination with formaldehyde – neatly transforming the product of their showcase reaction into a challenging target.
Paul Docherty is a science writer and blogger based in Reading, UK












No comments yet