Despite decades of work to develop alternative ways to make ammonia, the Haber–Bosch process is here to stay, Mark Peplow discovers
Despite decades of work to develop alternative ways to make ammonia, the Haber–Bosch process is here to stay. Mark Peplow discovers why the dinitrogen bond is so tough to break

Shining pipes intertwine like metallic tree roots, linking bulky high-pressure vessels that stretch 20m or more into the sky. Tonnes of hydrogen and nitrogen pulse through these throbbing arteries before uniting at scorching temperatures to make ammonia. You might catch a whiff of the pungent gas in the compressor house, but ammonia’s eye-watering odour is otherwise absent – after all, leaks are dangerous and uneconomical. Covering an area the size of two soccer fields, the gleaming plant can easily produce 2000 tonnes of ammonia each day. Welcome to the modern day Haber–Bosch process.
Rewind 100 years, and the first Haber–Bosch plant in Oppau, Germany, was about to begin producing ammonia at the rather more modest rate of 20 tonnes per day. But from that seed grew the most influential chemical process in the world. Offering ready access to synthetic fertilisers, it revolutionised agriculture and fed the 20th century’s population explosion. It also equipped armies with powerful explosives that have killed tens of millions; and it has left a legacy of nitrogen pollution that harms human health and destroys ecosystems by feeding algal blooms.
The Haber–Bosch process now makes roughly the same amount of ammonia as the sum total of all biological nitrogen fixation. ‘Nitrogen fixation is one of the fundamental reactions of nature,’ says Felix Tuczek of Christian Albrechts University in Kiel, Germany. ‘Every single nitrogen atom in your body has either gone through the Haber–Bosch process, or through biological nitrogen fixation.’
Despite its global impact, the fundamentals of the Haber–Bosch process have hardly changed at all. It still relies on high pressures of 150 to 350 atmospheres, temperatures between 350°C and 550°C, and (usually) an iron catalyst with a dash of potassium oxide on an alumina support.
Researchers had once hoped to develop a milder synthesis by mimicking the nitrogenase enzyme, which serenely fixes nitrogen from the air at ambient temperatures and pressures to feed leguminous plants. Most scientists have now given that up as unrealistic – the nitrogenase mechanism is still hotly debated, and synthetic analogues cannot compete with the optimised Haber–Bosch process.
The effort has not been wasted, though. Along the way it has helped to unpick one of the most mysterious processes of life, and develop methods to create high-value molecules by cracking chemistry’s toughest nut: the dinitrogen triple bond.1
Under pressure
By the turn of the 20th century, the world’s imperial powers were utterly dependent on regular shipments of sodium nitrate from mineral deposits in Chile. As a fertiliser, nitrate helped to feed their growing empires. And as a precursor to nitric acid, it helped to arm their soldiers with the latest explosives, such as trinitrotoluene (TNT). Breaking that dependency was the chemical challenge of the age. The key was to transform mere atmospheric ballast into something more reactive.
Working with Robert Le Rossignol, a young British scientist, the German chemist Fritz Haber developed a tabletop apparatus in 1909 that squeezed and heated nitrogen gas and hydrogen gas together to produce a steady stream of ammonia. But their catalysts – osmium or uranium – were unsuitable for a scaled-up version of the process. Carl Bosch and Alwin Mittasch of German chemical company BASF developed a cheaper iron catalyst, based on Swedish iron ore that contained traces of potassium ions. Its debut, when the Oppau plant opened in September 1913, marked the first example of industrial heterogeneous catalysis. By 1915, Bosch and Mittasch had also devised a process to convert ammonia into nitrate, which kept the German army well supplied with munitions. Haber and Bosch both won the Nobel prize in chemistry for their work, in 1918 and 1931 respectively.
Ertl’s work explained the background, but the basic kinetics had been around for years
Poul Erik Højlund Nielsen
The Haber–Bosch process now consumes 1–2% of the world’s energy output, and produced about 137 million tonnes of ammonia last year, according the US Geological Survey. Some 85% of that ammonia is used to make fertilisers. Roughly 2% of the world’s natural gas output is tapped to make hydrogen for the process, by steam-reforming methane at around 1000°C over a nickel catalyst to make carbon monoxide and hydrogen.
For such an important bit of chemistry, it has taken a remarkably long time to work out exactly what goes on inside an ammonia reactor. Practical details are well established – the catalyst starts off as magnetite (Fe3O4) before being reduced to porous iron by a stream of hot hydrogen, for example. But the catalytic mechanism itself was only figured out in the late 1970s and 1980s, largely by Gerhardt Ertl at the Fritz Haber Institute in Berlin, Germany, and Gabor Somorjai at the University of California, Berkeley, in the US.
Kinetic studies had shown that the rate-limiting step involved adsorbing and breaking apart nitrogen molecules. Ertl used surface chemistry techniques, such as photoelectron spectroscopy, to prove that the gas was cleaved into atomic nitrogen on the catalyst’s surface. He also revealed that potassium embedded on the catalyst’s surface helps neighbouring iron atoms to push electrons in to dinitrogen’s antibonding p-orbitals, which weakens the triple bond.
It was a triumph of catalytic detective work, but it did not have significant practical implications for the industry, says Poul Erik Højlund Nielsen, a senior scientist at Danish chemical engineering company Haldor Topsøe, which supplies the technology for more than half the world’s ammonia plants. ‘Ertl’s work explained the background, but the basic kinetics had been around for years,’ says Nielsen. Indeed, ‘amazingly little’ of Haber’s original chemistry has changed since Oppau, he adds.
Skillful engineering has made the process much more efficient, however, says Ib Dybkjær, a senior specialist in process engineering at Haldor Topsøe. Integrating the steam reformation with the ammonia synthesis created a continuous process that recycles waste heat. Topsøe estimates that such refinements have halved the energy required to produce a tonne of ammonia – to about 0.85MJ – since the 1950s. With an efficiency of around 70%, the modern process is close to its theoretical optimum, and industrial R&D is now focused on increasing the capacity and longevity of the plants, says Dybkjær. The world’s largest plant, built by German engineering firm Uhde in Saudi Arabia and completed in 2011, has a capacity of 3300 tonnes per day.
From plant to plants
Efficient as it is, the Haber–Bosch process undeniably consumes vast amounts of energy, and some researchers argue that even tiny improvements in catalyst efficiency could yield big savings. Others have hoped for a more fundamental shift – finding a catalyst that emulates nature’s ability to fix nitrogen gently from the air.
The industrial process relies on a chemical hammer-blow to cleave the bonds of the feedstock gases in one swift stroke, littering the catalyst’s surface with nitrogen and hydrogen atoms that rapidly combine to make ammonia. In contrast, nature uses a stream of protons and electrons to unpick dinitrogen one bond at a time – a surgical procedure orchestrated by nitrogenase, an enzyme found in microbes such as soil-dwelling Rhizobia.
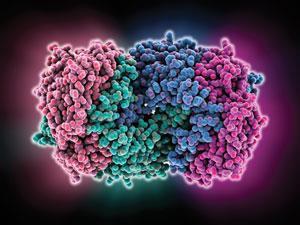
Nitrogenase has two major parts: an iron protein that acts as a delivery vehicle for electrons, and a molybdenum–iron protein that uses them to break dinitrogen apart. The electrons ultimately come from reduced ferredoxin, a powerful reducing agent generated during photosynthesis, while adenosine triphosphate (ATP) provides the energy.
Biological nitrogen fixation is far from being a paragon of efficiency. Turning each molecule of nitrogen into ammonia takes at least 16 molecules of ATP, and a quarter of the electrons involved in the reaction are ‘wasted’ to make hydrogen gas as a by-product. ‘These bacteria devote a lot of energy to it, which tells you how important nitrogen fixation is for life,’ says Patrick Holland at the University of Rochester in New York State, US.
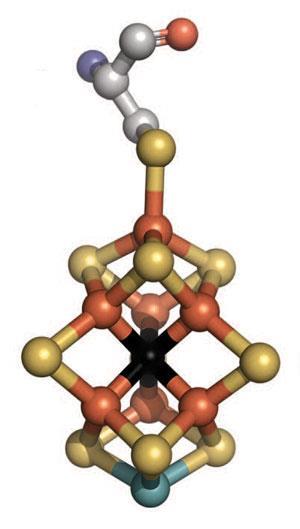
The catalytic heart of the enzyme is called the iron–molybdenum cofactor (FeMo-co), a cluster of seven iron ions, one molybdenum ion, and nine sulfides. Scientists are still unsure how it works – indeed, it was only in 2011 that x-ray crystallography definitively revealed a single carbon ion right in the middle of the cluster.2
Late last year, however, a quartet of US researchers – Brian Hoffman and Dmitriy Lukoyanov at Northwestern University in Illinois; Dennis Dean at Virginia Tech; and Lance Seefeldt at Utah State University – pulled together all that was known about the structure of the FeMo-co to propose a draft mechanism for the fiendishly complex reaction.3 ‘I often tell people that nitrogenase is proof of the existence of a mischievous god,’ laughs Hoffmann.
Based on a mixture of hard data and intuition, the team believes that a quartet of iron atoms in the FeMo-co forms the catalytically active site. The proposal is ‘absolutely not’ the final answer to the nitrogenase puzzle, says Hoffman, ‘but everybody who we have talked to seems to walk away saying “That makes sense”.’ He hopes that over the coming decades, evidence will prove or disprove each step of the mechanism.
Some are still doubtful that nitrogenase’s iron atoms bind and reduce dinitrogen. ‘It’s my view that molybdenum is where dinitrogen is reduced,’ says Richard Schrock of the Massachusetts Institute of Technology, US. ‘They’ve never really seen dinitrogen bound to these iron atoms.’
Schrock has good reason to think that molybdenum could do the job. In a landmark paper in Science in 2003,4 he and colleague Dmitry Yandulov showed that a molybdenum complex in solution could pepper dinitrogen with protons and electrons to produce ammonia catalytically. With a yield of around 65%, hopes were high that this synthetic system could be tweaked to match or even surpass nitrogenase.
And now? ‘Well, we haven’t done any better,’ Schrock sighs. ‘And we don’t have support for that work any more.’ Without specific grants, Schrock effectively stopped intensive work on the project about two years ago. He still has one researcher on the problem, and the project could spring back to life if they can improve their catalyst’s turnover in the next few months. ‘It’s not over till it’s over,’ he says. ‘But it’s getting near the end – for me at least – and it’s time for somebody else to step up and give it a try.’
This year’s top model
Along with a different molybdenum catalyst developed by Yoshiaki Nishibayashi of the University of Tokyo in 2010,5 Schrock’s complex still stands as a high-water mark in homogeneous, catalytic dinitrogen reduction. These soluble complexes are never going to replace the Haber–Bosch process, not least because they draw on huge molecules to supply protons and electrons, rather than cheap, compact dihydrogen. But they are useful as model complexes, says Hoffman, because they provide spectroscopic data that can be compared with nitrogenase to pin down the precise structure of reaction intermediates. With a thorough understanding of the mechanism, it may be possible to redesign nitrogenase to break other multiple bonds, he suggests, such as those in carbon dioxide.
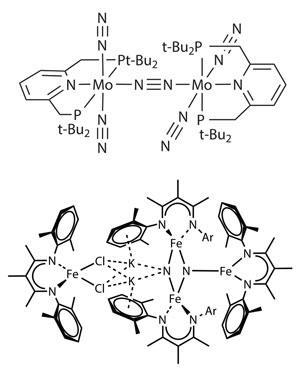
And compared with Haber–Bosch’s heterogeneous catalyst, soluble complexes generate intermediates that are much easier to trap and study, revealing how metals bind dinitrogen and break it apart, adds Holland. That in turn could provide more ways to functionalise dinitrogen, allowing synthetic chemists to tap an unlimited source for the nitrogen atoms in their high-value molecules. And it’s about time: a century after the Haber–Bosch process sprang in to action, ‘we still don’t have another industrial process that uses nitrogen as a feedstock’, says Michael Fryzuk at the University of British Columbia, Canada. ‘That’s a bit embarrassing for chemists.’
Dinitrogen is a poor ligand, and Fryzuk has spent years hunting for metal complexes that can hold on to it and then cleave the triple bond. A dinuclear tantalum complex,6 for example, can bind dinitrogen side-on to one metal atom and end-on to the other – this activated nitrogen will form new bonds with boron, silicon and aluminium. ‘But we’ve never been able to turn it over to make it catalytic,’ he says.
In 2011, Holland unveiled an iron complex that binds and cleaves dinitrogen, forming a structure containing bridging nitrides.7 Simply shaking this molecule with hydrogen in toluene forms ammonia at room temperature and pressure, and Holland is now testing other reactants to make more complex nitrogen derivatives. He acknowledges that it will be a huge challenge to make the process catalytic, though.
A big ship to turn
Despite the dominance of Mittasch’s iron catalyst, there are industrial alternatives. US engineering company KBR of Houston, Texas, offers a ruthenium catalyst that is 10 to 20 times more active than iron, and allows the process to operate at lower pressures of about 90 atmospheres, which reduces engineering costs. The catalyst itself is more expensive, however, which has limited its use to a handful of plants.
I don’t think there would be enough ruthenium to replace existing plant capacity
Poul Erik Højlund Nielsen
Aside from cost, ruthenium catalysts are also prone to hydrogen poisoning – the gas binds to the potassium oxide promoter, which can interfere with nitrogen adsorption. Michikazu Hara, Hideo Hosono and colleagues at the Tokyo Institute of Technology in Japan, last year unveiled an alternative promoter that suppresses hydrogen poisoning, effectively increasing the catalytic activity of each ruthenium atom by an order of magnitude. Their promoter is an electride, [Ca24Al28O64]4+(e–)4 – a crystal structure where some anions have been replaced by cavity-trapped electrons, which feed ruthenium nanoparticles embedded on the surface of the material.8
Nielsen agrees that it’s undoubtedly an improvement. But he points out that although Haldor Topsøe has also developed ruthenium catalysts, they have never improved the process enough to offset their higher cost. Ruthenium is also a scarce resource, with the world’s total reserves estimated at 5000 tonnes. ‘I don’t think there would be enough ruthenium’ to replace existing plant capacity, Nielsen says.
In 2000, Haldor Topsøe unveiled a new range of mixed metal nitride catalysts (including Fe3Mo3N, Co3Mo3N and Ni2Mo3N), which in tandem with caesium promoters were twice as active as conventional iron catalysts, yet much cheaper than ruthenium.9 Even these have not been commercialized, says Nielsen, because they are still at least ten times more expensive than the traditional iron catalyst.
Ultimately, most researchers agree that the biggest challenge to the Haber–Bosch process is likely to come from plant biology.10 Last year, for example, the John Innes Centre in Norwich, UK, took the lead in a £6.5 million project funded by the Bill and Melinda Gates Foundation to engineer cereal crops to associate with nitrogen-fixing bacteria, which might dramatically reduce the demand for fertilizer.
Until that happens, though, it will be business as usual for the Haber–Bosch process. And with today’s plants lasting for 30 years or more, industry is unlikely to make major changes to the iconic reaction in the coming decades, according to Dybkjær. ‘It’s a very big ship to turn around.’
Mark Peplow is a science journalist based in Cambridge, UK
References
1 S Hinrichsen et al, Annu. Rep. Prog. Chem., Sect. A: Inorg. Chem., 2012, 108, 17 (DOI: 10.1039/c2ic90033e)
2 K Lancaster et al, Science, 2011, 334, 974 (DOI: 10.1126/science.1206445)
3 B Hoffman et al, Acc. Chem. Res., 2013, 46, 587 (DOI: 10.1021/ar300267m)
4 D Yandulov and R. Schrock, Science, 2003, 301, 76 (DOI: 10.1126/science.1085326)
5 K Arashiba, Y Miyake and Y Nishibayashi, Nat. Chem., 2011, 3, 120 (DOI: 10.1038/NCHEM.906)
6 M Fryzuk, B MacKay and B Patrick, J. Am. Chem. Soc., 2003, 125, 3234 (DOI: 10.1021/ja034303f)
7 M Rodriguez et al, Science, 2011, 334, 780 (DOI: 10.1126/science.1211906)
8 M Kitano et al, Nat. Chem., 2012, 4, 934 (DOI: 10.1038/nchem.1476)
9 C Jacobsen, Chem. Commun., 2000, 1057 (DOI: 10.1039/B002930K)
10 P Beatty and A Good, Science, 2011, 333, 416 (DOI: 10.1126/science.1209467)










No comments yet