HIV protease inhibitors have been one of the big successes of rational drug design. Clare Sansom looks at the impact of structural biology on drug discovery
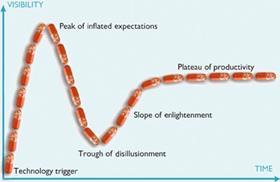
There are fashions in drug discovery, as in many fields of technical development, and these even follow common enough patterns to be mapped. Gartner’s ’hype cycle’ describes the path of a technology from initial adoption, over the peak of inflated expectation and through the consequent trough of disillusion, until reaching its ’plateau of productivity’, by which time it has evolved to fit its particular niche in the market and become stable. And structure-based drug discovery is a technique that has followed the cycle exactly. It was extremely fashionable during the 1980s, after the structures of the first important drug targets, including DNA and the enzyme dihydrofolate reductase, were published. Disillusionment set in during the following decade with the downgrading of these ’rational’ approaches in favour of high throughput screening and combinatorial chemistry. The pendulum has since swung back, and structural biology is now a mature science that has found its niche.
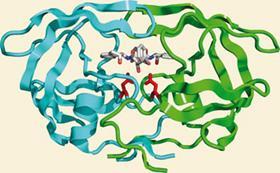
Yet it was during the years when this technique was so resolutely out of fashion that it scored perhaps its most stunning success to date: rationally designed HIV protease inhibitors. The first such drug, Roche’s saquinavir, entered the clinic in 1995 and, almost immediately, life expectancy for people with HIV and Aids began to improve. Only two years after saquinavir was licensed, Roy Gulick, a prominent physician at Cornell University, New York, US, involved in clinical trials of protease inhibitors, predicted that ’we may finally have the tools to turn HIV infection into a long-term, manageable and treatable disease, much like hypertension and diabetes’. Today, there are ten drugs in this class in clinical use - the latest, Tibotec’s darunavir, was licensed in 2006 - and they are key components of multi-drug HAART [highly active anti-retroviral therapy] treatments.
Rapid response
The global HIV pandemic is still less than 30 years old. The first cases of the disease that became Aids were reported to the US Centers for Disease Control and Prevention in 1981; four years later, not only had the disease agent been elucidated, but the complete genome sequence of that agent, the retrovirus later named HIV, had been determined. Scientists such as Steve Oroszlan, a researcher in viral oncology at the US National Cancer Institute, rapidly set to work on this new virus.
Oroszlan had proved that a similar virus, the murine leukaemia virus, matured by proteolytic cleavage - that is, as new virus capsules bud from an infected host cell, proteases slice up its payload of polyproteins into individual, functional proteins and enzymes. Oroszlan had also found that the protease cleavage sites were resistant to all known mammalian proteases, suggesting the presence of an essential retroviral protease.
The sequence of HIV revealed that it contained a similar protease to murine leukaemia virus. In 1985, Oroszlan published a paper showing, through site-directed mutagenesis, that protease was essential for HIV maturation and was hence a valid target for antiviral therapy. At a time when the medical community was struggling to cope with rapidly developing HIV epidemics, that was extremely welcome news.
A question of clan
Enzymologists have divided the proteases into broad ’clans’, divided by their mechanism of action and named according to the residues or chemical groups involved in that mechanism. The HIV protease sequence seemed most closely related to proteins in the aspartic protease clan, a group that includes the well-studied mammalian digestive enzyme pepsin. However, there were also significant differences. The sequences of pepsin and related enzymes are over 300 residues long and contain two copies of a characteristic tripeptide sequence necessary for enzyme activity: aspartic acid, threonine (or serine), and glycine - a trio generally referred to as DTG. In contrast, the retroviral protease is a much smaller enzyme - only 99 residues in HIV - and contains only one DTG sequence. It was clear from the beginning that this protein could not be active as a monomer.
Clan connections

The structural biology of the aspartic proteases has a very long history and pepsin in particular has a unique place in this. The first ever protein x-ray diffraction pattern was obtained by John Desmond Bernal and his student Dorothy Crowfoot [later Hodgkin] at the University of Cambridge from crystalline pepsin. This was published in Nature in 1934, and Bernal and Crowfoot wrote ’.now that a crystalline protein has been made to give x-ray photographs it is clear that we have the means of.arriving at far more detailed conclusions about protein structure than previous physical or chemical methods have been able to give,’ a prediction that has proved to be, if anything, an understatement. By the mid-1980s, atomic models of pepsin and several other aspartic proteases were known.
Mammalian aspartic proteases such as pepsin have a bilobal structure, with a large cleft in which the substrate binds and a flap that folds down over it when it is bound. The two characteristic tripeptides are symmetrically related and found at the bottom of the cleft, and the active site is supported by a tight network of hydrogen bonds that Tom Blundell and Laurence Pearl formally from Birkbeck College, University of London, UK, later termed a ’fireman’s grip’. One scientist who did much of the early work in this field was David Davies at the US National Institutes of Health, who remembers: ’some time in the late 70s I took wire models of the pepsin structure - all the models we had then - to a meeting where I was able to discuss it with Jordan Tang (who first sequenced the protein), Blundell, and others. We came up with the idea that there might have been an ancestral aspartic protease with a single DTG triplet that was active as a dimer, and that the mammalian proteases might have evolved through gene duplication. We didn’t realise then that enzymes with this structure could still be found in nature.’
By the late 80s, there were three groups working on crystal structures of retroviral proteases: Blundell’s, at Birkbeck College; Alex Wlodawer’s, at the US National Cancer Institute, Frederick, Maryland; and a group led by Manuel Navia at Merck, Sharp and Dohme in New Jersey, US. ’It was very difficult to get enough pure HIV protease to make crystals,’ remembers Wlodawer. ’Our first structural studies were on the related Rous sarcoma virus [RSV] protease, as that protein was more readily available. Our first crystal structure of the HIV enzyme used protein that Steve Kent, from California Institute of Technology, had produced by chemical synthesis.’
Pearl was a postdoctoral researcher at the Institute of Cancer Research (ICR) at Sutton in Surrey, UK, when the HIV sequence was published. ’After obtaining my PhD with Tom Blundell, I found myself in the fortunate position of knowing a great deal about aspartic protease structure and mechanism, and being in a lab doing state-of-the-art work in retrovirology - a group at the ICR had just discovered that CD4 was the receptor for HIV,’ he remembers. He studied alignments of the HIV sequence with other retroviral proteases, and noticed that ’everything needed for the aspartic protease mechanism was completely conserved in these sequences, but it was only half there’. He started to wonder whether these proteins could be the ’ancestral half-enzyme’ that Blundell, Tang and Davies had speculated about, and he worked with Willie Taylor at Birkbeck to build a structural model of a cut-down half protease. ’Our model could form a dimer with perfect two-fold symmetry,’ he says. ’Furthermore, we concluded that its mechanism would be similar enough to that of pepsin to be inhibited by known generic aspartic protease inhibitors, and that some of these might be good enough inhibitors to be leads for drug design.’ Blundell, who was independently thinking along the same lines, had started talking to collaborators at Pfizer in Sandwich, UK, about testing known aspartic protease inhibitors against HIV. ’Pfizer in the US had a series of compounds that inhibited aspartic proteases that are known targets for drugs against hypertension,’ he says. ’We were intrigued by the idea that an antihypertensive might be a lead compound for drugs against Aids.’
Structure racing
By 1989, the three groups were working against the clock, and against each other, to solve and publish their structures. The Merck group was the first to submit a manuscript on the HIV protease structure; they sent it to Nature , who sent it to Blundell to review. He realised that although the bulk of the structure was correct, the C-terminal part was inconsistent with the models that he had been building, and that his models seemed more plausible. ’However, knowing well how medically important this structure was, and believing it to be largely right, I recommended it for publication.’ He and Pearl laid out the experimental and predicted structures side by side in an accompanying News and Views article. The paper came out a week after the Wlodawer group had published the structure of the Rous sarcoma virus enzyme, and in the same week Irene Weber, working with Wlodawer, published a model of the HIV enzyme based on the RSV protease structure. When Wlodawer, and three months later, but to higher resolution, Blundell, published their x-ray structures, they proved that the Merck group had indeed traced the C-terminal end of the chain incorrectly.
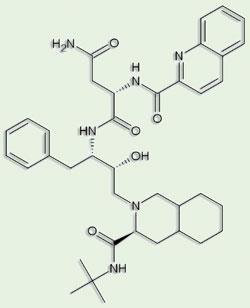
The enzyme has an extremely elegant, economical structure, made up largely of β-strands; it can preserve perfect two-fold symmetry if no substrate or inhibitor is bound. Yet the main interest in the structure had to be its potential role in the development of drugs against Aids. Polish crystallographer Mariusz Jaskolski, a long-distance member of Wlodawer’s protease group and first author on one of the structure papers, describes HIV protease as ’an outstandingly beautiful molecule from an outstandingly cruel virus’. Fortunately, the characterisation of a novel member of the already well studied aspartic protease family as a target gave the pharmaceutical industry a head start. ’Just about every pharma company had an HIV protease programme in the 1990s,’ remembers Blundell.
The first HIV protease inhibitors were so-called peptide mimetics, based on the structures of known peptide substrates of the enzyme, with alterations mainly to prevent cleavage of the target peptide bond, thereby ensuring the enzyme remained blocked. But peptides make poor drugs, and the lead compounds had to be modified to make them more ’drug-like’: essentially, smaller and less hydrophilic. Nevertheless, saquinavir was licensed for use against Aids a mere six years after the publication of the HIV protease structure - still a record for the time ’from bench to bedside’ for a novel drug. Pearl attributes part of this to the team effort of the groups involved, collaborators as well as competitors. ’While the individual contributions blur into history, this very tangible contribution to human welfare is something everyone involved should be immensely proud of.’
Reaching the plateau

By the time the first inhibitors entered the clinic, the pendulum had swung away from structure-based or ’rational’ drug design to the high throughput approach in which libraries containing thousands or even millions of unrelated compounds are screened against a potential drug target. It took another ten years, and the onset of disillusion as high throughput screening failed to increase the number or proportion of candidate drugs entering the clinic, for Wlodawer to see that his protease work had truly been ’a platform for the development of rational design strategies.’ And several of the original players in the HIV protease game have since made substantial contributions to the development of structure-based approaches that fit the twenty-first century. In 1999, Blundell co-founded Astex Therapeutics to exploit structure-based discovery methods; he is still a board member and scientific consultant for the company.
Blundell traces the thinking and method development that led to his involvement with Astex back to the HIV protease story. ’We soon began to realise that we had to screen non-peptide compounds to explore the whole possible range of protease inhibitors,’ he says. ’In fact, you need to explore as much of the available "chemical space" as possible. And as estimates of the number of potential "drug-like" molecules range from 1020 to a barely conceivable 10,200, even a library of a million compounds will only scratch the surface.’ The technology behind Astex involves screening with very small molecules or ’fragments’ rather than potential lead compounds; using structure-based methods, particularly x-ray crystallography, to identify fragment hits; and joining or growing the hits to form larger, more potent inhibitors.
Chris Murray, Astex’s vice-president of discovery technology, explains how this approach has built on both the structure-based methods of the 80s and the high throughput techniques of the 1990s. ’Structure-based drug discovery is all about using a small number of compounds in a clever way. High throughput screening is based on the premise that these clever approaches are unnecessary if enough diverse compounds can be tested. We have now come full circle; we are looking at compound quality as well as numbers. Our drug design methods use libraries of fragments that can be joined in many different ways, guided by structure and modelling, and they need both.’
Astex currently has four kinase inhibitors and one inhibitor of the heat shock protein Hsp90 in preclinical development or clinical trials for cancer. Interestingly, however, another of its programmes - in collaboration with a major pharmaceutical partner, AstraZeneca - concerns a novel aspartic protease: β-secretase, which cleaves amyloid precursor protein and is one of the key enzyme targets for Alzheimer’s disease. ’This is a membrane-bound, monomeric protease that resembles renin more than it does HIV protease, but has structural features that distinguish it from both. Tom [Blundell]’s accumulated decades of experience with aspartic proteases and their inhibitors have proved invaluable in this project’, says Murray. As the population ages, Alzheimer’s disease is becoming as high a research priority as Aids was twenty years ago. Will structure-based methods play as large a part in the discovery of drugs that halt the development, rather than just easing the symptoms, of this devastating condition, as they have in the HIV protease inhibitor story?
Clare Sansom is a freelance science writer based in London and Cambridge, UK
Additional information
A Wlodawer and J Vondrasek, Annu. Rev. Biophys. Biomol. Struct., 1998, 27, 249 (DOI: 10.1146/annurev.biophys.27.1.249)
A Mastrolorenzo et al, Curr. Med. Chem., 2007, 14, 2734
M Congreve, C W Murray and T L Blundell, Drug Discovery Today, 2005, 10, 895





No comments yet