Sarah Houlton finds out about some chemical tricks that can give a new drug the best possible odds of success
Sarah Houlton finds out about some chemical tricks that can give a new drug the best possible odds of success
By the time a new molecule achieves the status of potential drug candidate, there is a great deal riding on its success. The average cost of bringing a drug to market is $1 billion (?515 million) - that takes into account not only the direct cost of the discovery process, clinical trials and regulatory affairs, but the fact that so many of these precious molecules fail before they reach the market. Just 10 per cent make it.
’Attrition is diabolical,’ says Paul Leeson, head of chemistry at AstraZeneca Charnwood, in the UK. ’And the failure rate of candidate drugs is not getting better. It’s completely unacceptable.’
There is a depressing list of potential pitfalls: a drug may simply not work as well as had been hoped; it could cause severe side-effects; there might be difficulty in delivering it to the right site in the body; or an inability to create an acceptable dosage form. The list goes on.
’In the past five years there has been an increasing understanding of the cost of this attrition - particularly late on in the development process, and even once [products] have reached the market,’ says Tony Wood, head of discovery chemistry at Pfizer in Sandwich, UK. ’This has really heightened awareness of the need for medicinal chemists to achieve high selectivity and safety in the compounds we design.’

While one can never legislate for unexpected events in trials, there are tactics chemists can employ to ensure the compounds they put forward for development are more likely to succeed. ’The success or failure of a molecule is embodied in it when it’s made,’ Leeson says. ’We can make 1000 compounds - or more - in a discovery programme, and only a few of them are actually worth making.’
Wood agrees. ’If a compound is going to fail, it’s best to fail early - and better still before it’s even been made,’ he says. ’The moment a medicinal chemist writes a structure down on a piece of paper, or designs it on a computer, they fix all the problems the molecule will have. There are relatively few things you can do to alter its profile once it’s made - you can play around with salt forms or complex formulations, but other than that you get what you designed.’
Yet a small improvement in attrition would have a dramatic effect. ’It’s so bad that just a 5 or 10 per cent improvement could double the output of effective drugs,’ Leeson claims. ’Chemists have wasted too much time making compounds that have very little hope of succeeding. Often, the candidates we put through to development have some "baggage", but we should be far more critical and realistic about their prospects. Yet we may persist in the hope that apparently minor safety issues will go away. At best, development will be slow, and at the worst it fails. Why not try to make something better instead?’
Keep it simple
So what, in a chemistry sense, makes a good drug? And what makes a bad one? A decade ago, Chris Lipinski introduced the Rule of five which sets down the properties a molecule must have to increase its chances of becoming a drug. Yet even now, although the rules are known and understood by medicinal chemists worldwide, they are often disregarded. ’Because of the 10 year development cycle, there are only a handful of compounds now on the market whose development followed Lipinski’s rules, but it’s disappointing that a lot of current medicinal chemists don’t follow them either,’ Leeson says.
Lipinski’s Rule of Five
For a molecule to be drug-like, it should have:
- No more than five hydrogen bond donors
- No more than 10 hydrogen bond acceptors
- A molecular weight under 500
- A partition coefficient log P (a measure of lipophilicity) of less than 5
The understanding of what makes a molecule selective has greatly improved in the past five years. It’s not as simple as how well they bind to the target - a lot of it is related to how lipophilic (oil-loving) they are. Molecules are predominantly constructed from carbon atoms, but in the chase to improve selectivity by adding more interactions with the receptor, they become bigger, and because this adds carbon atoms, they are also likely to be more lipophilic. ’This makes them more prone to attrition,’ Wood explains. ’In the discovery and preclinical phases, molecules are often larger and more lipophilic, but by the time they reach clinical trials, those that have succeeded are likely to be smaller. One explanation is that by making a compound larger to increase its affinity for a target, you also make it more lipophilic, and thus less stable in an aqueous environment.
’This is where the extra potency really comes from - [the molecule] is driven out of the water and onto the receptor. And it’s a non-specific effect - it will be driven onto other receptors, too. That counters the positive influence of complexity increasing selectivity. Additionally, everything you gain in in vitro potency - which is, essentially, a ligand interaction - is undone by safety and pharmacokinetic problems that result once you get into the body.’
Lipophilic compounds are often more toxic, and have higher volumes of distribution, which means they will access more areas of the body, such as the central nervous system. According to Wood, it’s unknown whether there is a correlation between toxicity and free drug (the unattached molecules that float around the body) or bound drug (where it is attached to a receptor, plasma proteins or tissue). ’We do know that free drug drives efficacy in most cases,’ he says. ’For lipophilic compounds, there are usually much higher concentrations of bound drug than free drug, and so toxicity will be more likely if it is determined by total drug concentrations.’
Checking the solubility of your compound is another way to increase its chances of success. ’I can’t think of many good reasons for making an insoluble compound,’ says Leeson. ’There may be the odd specialist topical treatment where a level of insolubility might be good, but not in oral projects. Yet we’re still making a lot of insoluble compounds, and if they’d been looked at properly first, it would have been clear they were likely to be insoluble.’
But chemists are getting better at predicting solubility; Leeson says the high, medium or low solubility predictions are right about 80 per cent of the time. ’It’s worth working just in that 80 per cent space, taking the risk with the remaining 20 per cent of compounds that a good one will be disregarded. Of course there will still be some poor compounds, because the models are not perfect, but there will be far fewer.’ And, importantly, every compound generates more data to feed back into the predictive models, which improves them, too.
Ligand efficiency is another useful tool. This is a measure of the contribution of each of the heavy (non-hydrogen) atoms in a molecule to its affinity for the target, measured as free energy. ’If it’s above about 0.35, then you’re in good shape as you should be able to find a compound that is active at nanomolar concentration, with a molecular weight of about 500, within the Rule of 5,’ Wood says. ’Below that, you are more likely to have to break the Rule of 5 to get the affinity you need, which means your compounds aren’t going to be very efficient.’
Predicting the future
As chemists become better at solving existing problems, new issues rear their heads. ’About a decade ago, we became better at designing compounds that would have better pharmacokinetics,’ says Wood. ’The next problem was the observation that basic drugs like terfenadine [a hay-fever drug that was withdrawn] caused toxic cardiac effects, associated with the hERG, or human ether-a-go-go related gene.’
hERG is, essentially, an aqueous pore ion channel, and to bind to it, a compound merely has to occupy its large intracellular hole. This means that many different molecules bind to it, and because not many of the pores need to be filled to have an effect, it is very easy to activate it inadvertently. Careful structure-activity relationship studies are needed if it is not going to cause problems, for example paying particular attention to factors that are known to disfavour hERG activity, such as placing polar groups on the ends of the molecules.
Another issue that’s chemically driven is the formation of reactive metabolites. Again, it’s intrinsic to the chemistry and sometimes spotting what can go wrong in advance is difficult; often our knowledge of the chemistry isn’t good enough to anticipate that a compound is likely to be metabolised to a damaging unwanted species. But it’s important to consider it early on. ’If there’s a core element of the structure that might cause problems, it’s important to know that before many compounds based on it are made,’ Leeson says.
Case study - making drugs smaller
It’s normal for a medicinal chemist to take their hit compounds from high-throughput screening and increase selectivity and potency by adding molecular weight, and usually complexity and lipophilicity too. Wood’s team at Pfizer managed to reverse that trend in their non-nucleoside reverse transcriptase anti-HIV programme. ’It’s a challenging target site as it’s lipophilic, and the compound had to be active against several different mutants of the virus, so it had to be optimised against three or four different binding sites.’
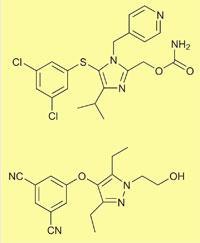
Capravirine (top) and UK-453,061 |
By focusing on ligand efficiency to design the compound, they showed that molecular weight doesn’t have to increase to get good efficacy and binding. ’The starting point was the existing experimental drug capravirine,’ he says. ’We chopped off the 4-pyridylmethyl group, which is known to cause problems with drug metabolism, and changed an aromatic dichloro function using protein x-ray guided design to replace it with a nitrile which is more polar, and achieved a similar interaction. We also changed some of capravirine’s hydrogen bonding groups to make them more flexible and capable of forming hydrogen bonds with more of the different virus mutants. Its carbamate became a hydroxyethylene group, which has more conformational freedom.’
The result was UK-453,061. ’We knocked about 100 off the molecular weight of capravirine, and also reduced its lipophilicity. We thought very carefully about the lipophilicity we added to the compound, and did it in the most effective way we could.’
Wood adds that it is becoming easier to predict which compounds will be unstable in the liver. ’Lipophilicity is important here, too, but various other functional group interactions are also involved. By building computational models that include many thousands of molecules and their activities, with their structural elements highlighted, we can see the relationship between the structural elements and stability. This is usually quite accurate, so it is now far rarer for us to make unstable compounds.’
Certain functional groups have been identified that might cause toxic problems. ’Anilines are a good example; they are easily oxidised and thus can covalently bond to proteins,’ Wood says. ’If you avoid anilines, you will decrease the likelihood of a late-stage failure. Others that can cause similar problems include Michael acceptors, aminothiazoles and thiophenes - anything that can be oxidised to an electrophilic group. But it’s also important to remember that there’s a very good relationship between total dose size and unexpected toxicity, so even if there is a "dangerous" group, if the dose can be kept below about 10mg the chances of problems are low.’
Knowledge is power
As chemistry data becomes increasingly abundant, available and organised, medicinal chemists get better at designing their compounds. ’Knowledge management techniques are very important,’ Wood says. ’We have access to vast quantities of data, and use complex statistical analysis to pull out knowledge and trends. In the past, we spent many hours in the library. Now we can code the literature into databases, so we can look at the whole medicinal chemistry literature for something that might work, at the press of a button.’

It’s in the hands of chemists to speed up the race from the lab to the cllinic © IMAGES.COM / CORBIS |
Leeson agrees. ’Whenever we make a molecule now, there are a dozen of pieces of information next to it - biological and pharmacokinetic profiles, safety data and so on.’
’Two things will have a dramatic effect on productivity: improving survival and increasing speed,’ Wood concludes. ’Speeding up the design-test cycle gets us so far, but we still move samples from one lab to another, isolate compounds and screen them. Better computer techniques mean we would need fewer iterations to get from first hit to final drug candidate; or each cycle could be done more quickly and all in one place. In future, we could see flow chemistry used to overcome this - a chemist designs a molecule, and a machine makes and tests it without isolating it. But that’s some way off in terms of the breadth of capability that would be needed.
’It’s not all about writing a molecule down on paper, though - it’s also the challenge of synthesising it as soon as possible. Good medicinal chemistry is enabled by good synthetic organic chemistry. And a good medicinal chemist will not only be able to design a compound that looks good, but one which doesn’t pose huge development challenges in synthesis.’ Speeding up the path from the lab to the clinic and reducing attrition in drug development pipelines are both in the hands of chemists - as long as they exploit predictive chemistry techniques, and design effective syntheses.
Sarah Houlton is a freelance science journalist based in London, UK
References






No comments yet