Being able to break selected C-H bonds empowers the synthetic chemist and could revolutionise the petrochemical industry, as Emma Davies discovers
Being able to break selected C-H bonds empowers the synthetic chemist and could revolutionise the petrochemical industry, as Emma Davies discovers
The pharmaceutical industry’s molecule makers can scarcely contain their delight at a new breed of catalysts that selectively and predictably attack carbon-hydrogen bonds in complex molecules. C-H activation catalysts can make life in the lab far simpler and greener - potentially cutting out a lot of reaction steps and so reducing solvent use. And it’s not just pharmaceuticals that can benefit - for years the petrochemical industry has been quietly hopeful that C-H activation catalysts will one day revolutionise petrochemical and fuel production. It all sounds so promising and yet while industry is happy to invest in academic projects, government funding bodies are repeatedly accused of failing to fund basic research in the field.
For simple molecules, and in ideal lab conditions, C-H activation can be quite straightforward. Classical methods involve removing the hydrogen atom using a base, a free radical, or an electrophile, usually promoted by a functional group that is adjacent to the targeted C-H bond. More recently, C-H bonds at positions more remote from functional groups have been catalytically activated by transition metals such as platinum, palladium, rhodium, and iridium. Homogeneous catalysts follow several mechanisms, but the metal first coordinates to the C-H bond before cleaving the bond to generate discrete M-C bonds.
But the pharmaceutical industry has the problem that it is notoriously difficult to target selected C-H bonds in its complex and polyfunctional molecules. Some success has been achieved through activating C-H bonds with adjacent heteroatoms or pi systems, or by using substrate directing groups.

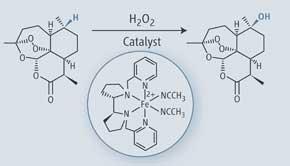
However, in 2007, Christina White and Mark Chen at the University of Illinois, US, achieved what many would never have been thought possible as little as a decade ago - developing an iron catalyst that selectively and predictably oxidises C-H bonds in complex molecules to give COH.
’What’s unique about our research is that we were able to oxidise very strong, unactivated C-H bonds - we didn’t need any directing groups,’ explains White. ’We think that we have been able to do that because there are actually subtle electronic differences between C-H bonds in a molecule. There are also very subtle steric differences depending on where the bonds are. The catalyst that we developed - which we coined Fe-PDP - is able to distinguish these differences.’
The researchers successfully demonstrated their new catalyst on several complex products, including the antimalarial artemisinin. ’We were able to distinguish one among five tertiary C-H bonds in artemisinin based on electronic properties,’ says White. ’For a long time people didn’t believe that that kind of selectivity was possible, especially not in a very predictable way.’
Selectively targeting C-H bonds in complex molecules can significantly streamline the molecule making process. ’When organic chemists make molecules, they often try to have all the functionality in the molecule at the beginning and then they carry it through the synthesis, which requires a lot of unwanted protection and de-protection steps,’ explains White. Being able to introduce oxygen at a very late stage takes away the need to protect and de-protect it. ’If you can put oxygen in right at the end - predictably and with high selectivity - then it can be very empowering. It can save you a lot of steps in the synthesis and make it much more efficient. We have many examples demonstrating the streamlining effect of applying a late-stage C-H oxidation strategy,’ enthuses White.
And it’s not just oxygen that can be inserted. ’Potentially you could also insert nitrogen or carbon at a late stage. You can make a lot of very significant modifications to the molecule’s core,’ says White. When chemists do late stage diversifications on molecules they typically replace one functional group with another. ’This chemistry can allow you to change the hydrocarbon core to add functionality,’ says White.
White’s Fe-PDP catalyst will soon be commercially available from US-based chemicals manufacturer and supplier Strem. ’It’s something that anyone can run very easily,’ says White. ’We’re really excited about getting people to use the catalyst. We’ll get a lot of feedback in terms of what works and what doesn’t - it’s a very exciting time.’ Predictability is key for White. ’Without predictable selectivity, no one would dare to use these methods towards the end of their synthesis,’ she says.
White and her team are also ’tweaking’ their iron catalyst and looking at other transition metals. ’We also have a palladium system with bis sulfoxide [bis-sulfoxide/Pd(OAc)2] that does very selective and predictable allylic C-H oxidation chemistry. It can do allylic oxidation, amination, and even alkylation chemistry,’ she explains. In fact, it did the first ever Pd(II ) catalysed allylic C-H alkylation and provides a new way to make C-C bonds directly from C-H bonds.
White’s group is just one of a number looking to tackle C-H activation in complex substrates. ’White falls into a group of a large number of young people who are interested in C-H activation and doing incredible things with it,’ says Robert Bergman, at the University of California, Berkeley, whose seminal work on C-H activation really got the field going.
C-H activation: the early years

The first C-H activation by a transition metal was reported in 1965 by Joseph Chatt at the University of Sussex, UK, who used the extremely electron-rich species Ru(dmpe)2 to insert a ruthenium atom into a C-H bond of naphthalene. Several other examples were subsequently discovered in which apparently electron-rich metal centres were found capable of oxidatively adding to C-H bonds, but essentially all of them involved intramolecular reactions or aromatic C-H bonds. Then a Russian chemist called Alexander Shilov did a series of highly important experiments and discovered some of the first intermolecular homogeneous metal-catalysed alkane functionalisation reactions.

Robert Crabtree did his PhD with Chatt and went on to provide important examples of alkane oxidation. ’One tea-time somebody asked Chatt what would be the next project if the problem of nitrogen fixation - how to convert nitrogen to ammonia - was solved. He said that it would be C-H activation. As an impressionable young graduate student I made a mental note of this,’ recalls Crabtree. ’When I came to the US, Roald Hoffman told me that I would have to do something pretty spectacular to get tenure at Yale,’ says Crabtree. ’So I tore up all of my previous plans and arrived at Yale in 1977 with the plans of C-H activation in my pocket, so to speak.’ Crabtree and his graduate student set to work studying alkane dehydrogenation by iridium complexes. ’We got a very early hit - it was beginner’s luck probably,’ says Crabtree. In 1979, he reported that [(Me2CO)2IrH2(PPh3)2]+ dehydrogenated cyclopentane and cyclooctane to give the corresponding cycloalkadiene iridium complexes.
Then in 1982, two groups made the first direct observations of intermolecular alkane oxidative addition, reporting that iridium from an organometallic complex could be inserted between the atoms of C-H bonds in saturated hydrocarbons such as cyclohexane and neopentane to give stable alkyl iridium hydrides. Robert Bergman at the University of California, Berkeley, US, led one of these groups. ’The key to what we found was the fact that we were monitoring our reactions using NMR spectroscopy,’ recalls Bergman. ’The C-H activated products were pretty sensitive. If we had been using the standard purification procedures that were common at the time, we probably wouldn’t have been able to isolate and fully characterise them.’

The researchers irradiated an iridum dihydride complex with various solvents in sealed tubes and monitored the reactions. Changing the solvent gave a different chemical shift for the hydride absorption. ’This essentially told us that alkanes like cyclohexane and pentane, and ultimately methane, were forming stable products,’ says Bergman. He was quick to grasp the significance of the result and published it as soon as possible.
Bergman has spent 30 years studying the characteristics of C-H activation, its reaction mechanisms and its application to organic synthesis problems. He would dearly love to isolate and fully characterise an alkane C-H activation complex, formed before the oxidative addition of a C-H bond. ’There is good evidence that they are detectable. We’re trying to synthesise an isolable alkane complex - where the alkane is just coordinated to the metal through a C-H bond. At some point somebody will find the right set of metals and ligands to isolate one of these things and do a crystal structure on it. And it would be nice if we could do that.’
R H Crabtree, J M Mihelcic, and J M Quirk, J. Am. Chem. Soc., 1979, 101, 7738
A H Janowicz and R G Bergman, J. Am. Chem. Soc. , 1982, 104, 352
Pharma’s friend
It’s easy to see why the pharmaceutical industry is so enamoured with C-H activation catalysts. Many of the industry’s discovery chemists have already tried C-H activation catalysts to help build new molecules in the lab. ’For the pharmaceutical industry, one of the key drivers is to reduce the number of steps in processes - that has a clear cost element and a clear speed element. Catalysis underpins this driver and is very attractive to us,’ says David Hollinshead, science policy director at AstraZeneca.
’C-H activation is probably one of the hottest fields right now in synthetic chemistry,’ adds Carl Decicco, senior vice-president of discovery chemistry at pharma firm Bristol-Myers Squibb (BMS). ’The types of C-H bonds that White is activating are the ones that everyone would like to go after. Being able to selectively go down a carbon chain and activate a hydrogen is a dream scenario.’
Steve Brickner, who researches anti-bacterials at Pfizer and collaborates with White’s group, is particularly impressed by the specificity with which highly complex molecular frameworks can be modified. ’This specificity could previously never have been envisioned outside the world of enzymatic manipulation,’ says Brickner. ’I also see considerable attraction in using this chemistry to generate metabolites or pseudo-metabolites of drugs or drug candidates in an orderly and predictable fashion,’ he adds. ’Another extremely attractive feature may be the ability to significantly simplify the synthesis, on scale, of certain key intermediates required for drugs.’
It’s a gas
The pharmaceutical industry can easily reap the rewards of C-H activation chemistry, particularly in discovery chemistry. But it’s tougher for the petrochemical industry, which spends vast amounts of money constructing plants designed to run very specific reactions, and so needs to be presented with a very strong business case to depart from current methods. C-H activation is competing with highly efficient processes, albeit energy intensive ones. But researchers are convinced that C-H activation holds great promise and will allow the vast reserves of natural gas (composed mainly of methane) to be used as an alternative to oil as a feedstock for fuels and chemicals.

Current processes for breaking alkane C-H bonds are costly - needing temperatures above 500?C and consuming vast amounts of energy. Catalytic C-H activation has the potential to dramatically lower the temperatures required and could lead to a ’new paradigm in energy and materials technology in the 21st century,’ says Roy Periana, director of the Scripps Energy Laboratories, the Scripps Research Institute, Florida, US.
One important and realistic application of C-H activation could be in converting natural gas to liquid products. ’It boils down to the problem of converting methane economically to liquids, which can be shipped cheaply or used as a cheap precursor to olefins [alkenes] etc. If it can be done cheaply then natural gas can replace oil,’ explains Periana.
Methane is currently heated with water at 900?C to produce syngas - a mixture of carbon monoxide and hydrogen - which in turn can be converted to methanol or directly to fuels. But with the appropriate catalyst, methane can be converted directly to liquids such as methanol at far lower temperatures and lower costs. Developing such a catalyst is a tough task. Unlike pharmaceutical researchers, Periana’s team is restricted to using air as the oxidant and has to compete with an industrial reaction that gives methanol in 70 per cent yield. His team has already made groundbreaking advances, receiving international acclaim for a set of C-H activation catalysts that convert methane to methanol. The second of these, developed in 1998, is a platinum(II )bipyrimidine catalyst that, in the presence of concentrated sulfuric acid, converts methane into methanol - via a methyl bisulfate intermediate - in 70 per cent yield.
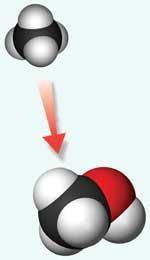
But the slow rates of these catalysts means that they still can’t compete economically with existing conversion processes. ’That’s the focus of our efforts - to get the rates of reaction fast enough with methane so that we can reduce the overall cost of the process,’ says Periana.
He remains hugely excited about C-H activation. ’The scope of the reaction is very broad. For example, if we can insert oxygen atoms in ethane as we have done for methane we could even replace the current multi-step ethane to ethylene glycol [ethane-1,2-diol] conversion, which is worth $20 billion per annum,’ he says.
Periana is also trying to fulfil his ambition of using C-H activation catalysts to develop low temperature methane fuel cells, where the methane releases hydrogen right at the anode. ’There is no fuel cell today that will "burn" methane efficiently at 200?C. An exciting application of C-H activation would be to develop anodic catalysts that could "tetra hydroxylate" methane at temperatures below 200?C. The resulting hypothetical molecule, "tetrahydroxymethane," is effectively the same as CO2 and water.’ Mediating such a reaction at the anode would allow the conversion of methane and two liquid water molecules to CO2 and hydrogen at temperatures low enough to reduce costs and increase efficiency to almost double that of current fuel cells.
The downside of Periana’s existing catalysts is that they need high concentrations of sulfuric acid. Diluting the acid with water for fuel cell use lowers the rate at which the catalyst works. ’The current catalysts are 1000 times too slow,’ reveals Periana. He has decided to take the drastic step of ’a complete redesign’ of his catalysts. ’We think we understand what happens when we add water and are redesigning our catalysts so that water is no longer an issue.’
Many systems already do C-H activation in water but the reactions are too slow to be practical, and most can only convert the C-H bond to metal-carbon bonds. ’We can’t sell the M-C bond - we’ll have to convert it to COH or some other useful functionality, reversibly and repeatedly,’ says Periana. It will be worth the extra effort. ’If we can commercialise the new hydroxylation chemistry it will revolutionise the petrochemical industry,’ he says.
Periana is to be applauded for tackling head-on the problem of slow catalyst rates. C-H activation catalysts are failing to be adopted in large-scale industrial process because of the high loadings required. ’The entire field is facing the problem that none of the current catalysts are applicable to industry,’ says Robin Bedford, professor of chemistry at the University of Bristol, UK. The reactions that chemists are trying to replace, explains Bedford, such as the highly popular Suzuki cross-coupling reaction, can use catalyst loadings of parts per million. ’If you look at the catalyst loadings of all the aromatic C-H activation reactions, they tend to be in the percentage range at best. There are very few aromatic C-H activations where you can use catalyst loading of less than 1 per cent. Most, if not all, are hampered by a need to use relatively high catalyst loading.’
The problem lies in the fact that C-H activation tends to be slow and is often the rate-limiting step of a reaction. ’Meanwhile, the rate of catalyst decomposition versus the catalysis rate is relatively high,’ explains Bedford. The answer is to gain a deeper mechanistic understanding, he adds. ’There are several mechanisms for aromatic
C-H activation and it can change - very recent results suggest that very subtle changes to your catalyst or your reaction could lead to the rate-limiting step swapping from one
C-H activation process to another. We really need to pin it down before we start addressing how to optimise the catalysts.’
BMS’s Decicco agrees that the catalysts are not yet ready for the pharmaceutical process arena. ’But the chemistry is useful in the discovery setting - we have already used it,’ he says. ’It’s very rare that a discovery synthetic route is used in the process arena. Process chemists are really great at coming up with clever solutions to problems and we don’t usually worry too much about the final route in the discovery setting,’ he explains.
Back to basics
Despite the calls for more basic research, government funding for such projects on both sides of the Atlantic is getting harder to come by. ’There are still a lot of people in the funding agencies who believe in these things but they’re under a lot of pressure to come up with things that pay off right away,’ says C-H pioneer Bergman. Bedford gets almost all of his funding from the pharmaceutical industry and is frustrated that he has had very little responsive mode funding from the UK’s Engineering and Physical Sciences Research Council for his work on C-H activation.
Decicco feels very strongly about the funding issue: ’C-H activation is extremely important and it is clear to me that getting government funding for basic research is more difficult than it should be.
’If you listen to the great professors in chemistry, they all say the same thing - that fundamental and strong organic chemistry is the basis of many new discoveries,’ says Decicco. And Bergman agrees. ’I always thought that C-H activation was such a fundamental transformation that it was completely justifiable to study it, whether or not it turned into something practical,’ he says. ’I would never have given up on the field mainly because I thought it was chemically really interesting.’
’A lot of people thought it very unlikely that any useful catalytic chemistry would come out of C-H activation chemistry. The fact that there’s now lots of it is significant and means that people shouldn’t be too pessimistic,’ says Bergman. The younger generations of C-H activation researchers need to hold on to their enthusiasm and ride out the funding storm.
Emma Davies is a science writer based in Bishop’s Stortford, UK
References






No comments yet