There is much more to DNA than that elegant double helix. Philip Ball explores the twists and tangles of chromatin
There is much more to DNA than that elegant double helix. Philip Ball explores the twists and tangles of chromatin
Why isn’t my arm a leg, or my liver a kidney? Their cells all have the same genes. But genetics isn’t just about what genes you have - it also depends on how you use them. This crucial aspect of our biological identities has been overshadowed by a focus on the identity of our genes, and how these differ from those of other people and other species. But it has now become clear that genetics can’t be fully understood until we have a better picture, not just of what is in our genes, but of how they are labelled and packaged.
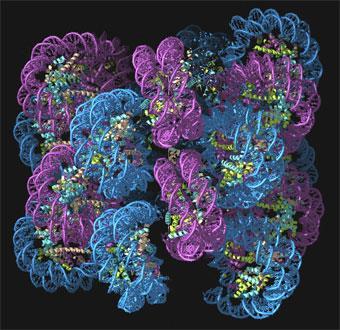
This evolving view of gene packaging is quite different from the classical image of genes as stretches of pristine double-stranded DNA. Our genetic book has been scribbled all over: there are marginal notes and sections crossed out. Not only do these textual changes determine what the genetic book says, but they reveal the book’s history, showing how its environment has modified and shaped its message.
Perhaps part of the reason for the relative neglect of this aspect of gene function is that it is a tremendously hard problem, to which the answer is likely to be messy. It could be said that molecular biologists have been in denial ever since Francis Crick and James Watson discovered the structure of DNA in 1953. That beautiful double helix, with its genetic information written into the spiral staircase of paired nucleic-acid bases, offers such an elegant picture of the chemical principles of life and inheritance that everyone fell for it.
This image of DNA is celebrated in the corridors of biology labs the world over. But when we come face to face with DNA in the cell, it’s like meeting a movie star whose airbrushed publicity photos don’t look at all like the real thing. You would barely recognise Crick and Watson’s perfectly-formed molecule in the tangled, twisted and bent spaghetti that is stuffed inside the nuclei of our cells.
It may be messy, but the packaging of DNA into the chromosomes seems to have a deep logic to it. These rules determine how available the DNA is for being read and transcribed into RNA - the first step in the conversion of genetic information to proteins. To understand why some genes are silent while others are actively transcribed - and thus what allows genetically identical cells to become differentiated into different tissue types - we need to unravel the packaging.
Wound up genes
In eukaryotic cells, where the DNA is housed in a cell nucleus, the chromosomes aren’t pure DNA. About half of their material is protein: chromosomes are made up of a fibrous composite of DNA and protein called chromatin. It now seems that this structure can determine the activity of genes, and that it therefore plays as big a role in the genetic make-up of organisms as the basic sequence of the genes themselves. The chemical nature of chromatin may hold the key to understanding how a set of genes creates a complex, multicelled organism like ourselves - and also to how this process sometimes goes awry and leads to conditions such as cancer.
In 1974, Roger Kornberg, then working at the UK Medical Research Council’s Laboratory of Molecular Biology (LMB) in Cambridge, proposed that chromatin is like a string of beads. The DNA double helix, he argued, is looped around barrel-like units made up of proteins called histones. Each of these barrels is made of two copies each of four different histone proteins - H2A, H2B, H3 and H4. Segments of DNA about 146 base-pairs long are spooled around each histone octamer in a double loop. Each of these DNA-histone units is called a nucleosome, and chromatin consists of a succession of nucleosomes linked, by short stretches of DNA, into a flexible chain. There is a fifth histone protein, called H1/H5, attached to the nucleosome at the places where the DNA first makes contact with, and exits from, the protein core.
The nucleosomes are themselves packed together to form chromatin fibres about 30nm wide - but no one knows quite how. One idea is that the fibres might be supercoiled into a solenoid, or one-start structure, as happens if you twist the helical cables of old telephones. In this structure, each pair of successive nucleosomes is connected by a bent piece of linking DNA. But it has also been suggested that adjacent nucelosomes along the chain could be connected by straight segments, creating twinned stacks of nucleosome helices - two-start structures.
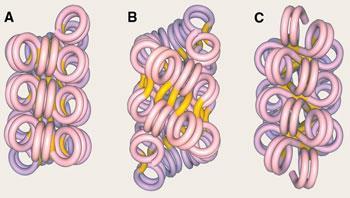
In 2004, Timothy Richmond’s research team at ETH in Zurich, Switzerland, argued that chromatin fibres must have a two-start form.1 But last year, Daniela Rhodes and her colleagues at the LMB challenged this idea.2 They chopped up and reconstituted chromatin with varying lengths of DNA linking the nucleosomes - from 10 to 70 base pairs (bp). They found that, whereas two-start structures should have a fibre width that increases gradually with increasing linker length, in fact the fibres fall into two distinct classes. For linkers of up to around 40 bp, the fibres have a diameter of 33nm and contain 11 nucleosomes in every 11nm of length. But with linkers of more than 50 bp, the fibres are 44nm wide and have 15 nucleosomes per 11nm length. They proposed that these results could be understood with yet another structure, in which nucleosomes in adjacent helical arrays are interdigitated.

The argument isn’t yet settled. Julien Mozziconacci of the Universit? Pierre et Marie Curie in Paris, France and his colleagues have tried to sort it out by combining Rhodes’ results with molecular modelling.3 They have built computer models of chromatin fibres that include every single atom, to see whether the proposed structures will fit without any atoms getting in each others’ way. They say that chromatin fibres can adopt many different structures, including the supercoiled solenoid and various two-start helices - meaning that almost all of the theories could be correct.
’We assumed that nucleosomes are stacked into columns which are, in turn, coiled to form 35 or 45nm fibres,’ says Mozziconacci. ’We identified 15 different ways to do this, depending on the number of columns and the handedness of their coiling, as well as that of the DNA path around the fibre axis.’
It all depends on how many base pairs there are between one nucleosome and the next, which can vary in different parts of the genome and under different conditions. In both the centres and the tips of chromosomes, for example, the average repeat length is different from the rest. The repeat length also seems to change when chromosomes become compact as a cell prepares to divide. To accommodate such changes, Mozziconacci thinks that the H1/H5 histone plays the role of an adjustable clip that alters the angle at which the DNA meets and exits from the nucleosome core, so as to make for a tidy fit in the packed-up fibre.
This shape-shifting nature could provide a mechanism for controlling gene function. ’I think that the histones evolved so that they can use the helical shape of DNA to acquire this polymorphism,’ says Mozziconacci. ’Shuffling of nucleosomes to alter the repeat length might be a way of regulating transcription and other chromosomal processes.’
It’s still an open question how all of this, which stems mostly from work on chromatin isolated in test tubes, carries over to living cells, but it seems clear that there’s much more to chromatin structure and packaging than the 30nm fibres. ’I believe there are definite higher-order structures of chromatin in the cell, above the level of these fibres,’ says Mozziconacci. ’I’d speculate that there are motifs such as kinks and crosslinkings which result in an ordered architecture.’
For one thing, there are in fact two types of chromatin in the nucleus of a eukaryotic cell, at least during the interphase period - when it is not about to divide. Most is in the form of euchromatin, a fairly open, gel-like fibrous tangle. The rest is heterochromatin, which is much denser and confined to a few small patches. Because DNA wound onto histones and packed tightly into fibres can’t be accessed by the molecular machinery that controls transcription of genetic material into RNA, there is a complex array of enzymes involved in unpacking and repacking (’remodelling’) chromatin, which is vital to the readout of our genes.
It’s tempting to imagine that euchromatin is unpacked and accessible for transcription, while heterochromatin is inactive, like a compressed data file. But it’s not that simple. A lot of the DNA in euchromatin never gets transcribed - so it’s not obvious why it should be left ’open’. Conversely, chromosomes containing a large amount of heterochromatin can be transcriptionally active. Some researchers think that euchromatin is actually just a blanket term that hides structural subtleties of chromatin we don’t understand. ’How heterochromatin and euchromatin structures differ is unclear,’ says chemical biologist Michael Grunstein of the University of California at Los Angeles.
To fully understand transcription - what determines if genes are ’on’ or ’off’ - we must therefore get to grips with what chromatin remodeling entails. It is done by various big protein complexes, which distort the contacts between histones and DNA or move nucleosomes around. One possibility is that these chromatin-remodelling complexes (CRCs) simply loosen up the chromatin so that nucleosomes can slide along the DNA chain. But it now seems likely that their activity is more specific. Last October, Gernot L?ngst of the University of Regensburg found that several CRCs position nucleosomes quite precisely by looking for specific DNA sequences.4 And since there are hundreds or even thousands of different CRCs in the nuclei of eukaryotic cells - at concentrations that may provide as many as one CRC for every ten or so nucleosomes - it seems inevitable that they must have rather specialised functions. Since this remodeling can alter the accessibility of genes, determining whether they are active or silent, it seems clear that CRCs must play a crucial role in defining a cell’s genetic status.
Read the label
Both remodeling of chromatin by CRCs and transcription of DNA are governed by chemical labels stuck to the histones or to the DNA itself. Specialised enzymes add or chop bits off the proteins or DNA, providing signals for other enzymes that indicate whether the genetic material should be read out or ignored. These modifications are a way of changing the activity of genes without changing the genes themselves. Such behaviour is called epigenetic. In effect, epigenetic processing enables cells to select from the complete genome just those genes that it needs to serve a particular function - which can change with time. Epigenetic modifications are reversible, so that a cell’s active genetic resources are being continually manipulated. Some researchers believe that epigenetic modifications of chromatin should be regarded as a kind of code that complements the basic genetic code of DNA, and is even capable of over-riding it.
Others say that code is not the right word - even if chemical alteration of histones can rearrange chromatic structure and turn genes on and off. ’A code needs to be consistent, predictive and combinatorial,’ says Grunstein. (Morse code is a true code in this sense because, whereas one dot means ’e’, two dots mean ’i’ rather than ’ee’.) ’Epigenetic modifications are none of these.’
It seems like a semantic point, but at its core is the question of how information is encoded in the working genome: a true epigenetic code would be more than just a series of chemical on-off switches, but would in some ways be as fundamental as the genetic code of DNA base pairs - a part of the language of life.
Geneticist Bryan Turner of the University of Birmingham, UK, who first proposed it,5,6 admits that the idea of an epigenetic code ’is still very controversial’. ’We do not yet have the finding that will prove or disprove the epigenetic code hypothesis,’ he says. ’Histone modifications may be involved in all sorts of processes. If there is a heritable code, then we need to clear away modifications due to all these other processes before we can find it.’
Whether it’s a code or not, the repertoire of histone modifications is beginning to be catalogued. One of the most common changes is acetylation - where an acetyl group is substituted in place of a hydrogen atom on the amino group of a lysine residue. This can occur on several of the lysines of all four core histones, particularly H3 and H4, so some nucleosomes can be multiply acetylated. Acetylation of lysine removes its positive charge, reducing the tendency of the histone to stick to negatively charged DNA. That in turn can open up the chromatin structure and activate genes for transcription.
Enzymes that add a methyl group to arginine and lysine residues - the process of methylation - can be responsible for either activating or silencing genes. Transcriptionally silent heterochromatin often contains nucleosomes in which histone H3 has been methylated at a site to which the enzyme HP1, which is involved in repression of genes and packaging of heterochromatin, binds. It was once thought that methylation is irreversible, but in 2004 a team at Harvard Medical School in the US discovered an enzyme that demethylates lysine.7 Several others are now known.
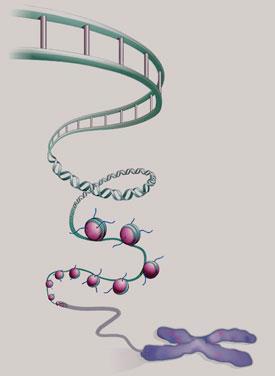
Other histone ’marks’ include phosphorylation, ubiquitination (attachment of the small protein ubiquitin, usually used as a marker of old or damaged proteins that need chewing up by proteasomes) and citrullination (the transformation of arginine into the amino acid citrulline). Individual nucleosomes may contain several marks at once. Last year, Bradley Bernstein of the Massachusetts General Hospital in Boston, US, and his coworkers reported that developmental genes in mouse stem cells carry both ’activating’ and ’silencing’ histone marks.8 This, the researchers think, is a way of fine-tuning the genes during the developmental process, for example keeping them inactive but poised to go into action once the stem cells become committed to developing into a specific tissue type.
Some epigenetic marks are associated with cancer cells. For example, Grunstein and his former student Siavash Kurdistani have found that certain patterns of acetylation and demethylation of residues in the H3 and H4 histones seem to appear in men with prostate cancer.9 A link between histone marks and cancer would be understandable, because these marks are often used to label damaged DNA so that it can be identified for repair by specialised enzymes.10 A break in the double helix can, for example, lead to phosphorylation of many H2 histones in that region within seconds, in effect putting up a distress flag for DNA-repair enzymes. Specific CRCs are thought to have a role in repairing such fractures. If the damage isn’t put right soon enough, however, that can lead to breakdowns in the cell cycle and the onset of cancer - which is why labels would be abundant in the chromatin of a person at risk of developing it. Researchers hope that these histone marks can therefore offer an early-warning system for prognoses of cancer in clinical screening.
Rewriting evolution
Perhaps most dramatically of all, it seems that some epigenetic changes can be inherited: a kind of Lamarckian inheritance of acquired characteristics called epigenetic inheritance, which undermines the classical rules of genetic inheritance. One way in which this can happen is by chemical marking of DNA itself, rather than histones. Between 2 and 7 per cent of the cytosine bases in our DNA (and as much as 30 per cent in some plants) have methyl groups attached, which alters the activity of the associated genes. Some DNA-methylating enzymes, such as Dnmt 1, tend to stick methyl groups onto CG-GC base pairs that already have them - and so methylation can be inherited by a replicated strand. In 2003, for example, a team at the University of Sydney, Australia, showed that mice could inherit a kinked tail caused by epigenetic methylation of the DNA in a gene called AxinFu.11 It’s also possible that histones attached to replicating DNA might pass on their modifications to those on the daughter strands.
In organisms that reproduce sexually, epigenetic modifications of chromatin are generally cleared during the merging of germline cells (sperm and egg) in the process of meiosis. Azim Surani of the University of Cambridge and his coworkers have argued recently that the stripping of methyl tags from DNA during this reprogramming may involve enzymes involved in DNA repair.12 But such wiping of epigenetic changes doesn’t always seem to happen - sometimes these modifications get passed on. The classic example is called paramutation, a type of gene silencing in which the gene variant (allele) acquired from one parent’s DNA affects the other allele in the progeny cell. First identified in plants in the 1950s, paramutation is still not well understood, but it is thought to involve epigenetic chemical modification of either DNA or histones in a way that is mediated by RNA.
’While it is clear that chromatin states are stable and can be preserved through cell division, the precise mechanistic roles played by the histone modifications themselves remain obscure,’ says Bernstein. All the same, there’s now no doubt that the neo-Darwininan picture of genetics and inheritance is incomplete, and we won’t fully understand either how evolution works or what makes us human until we have done some more unraveling of the subtle chemistry of chromatin.
Philip Ball is a science writer based in London, UK.
References
1 B Dorigo
10 J A Downs et al, Nature, 2007, 447, 951
11 V K Rakyan et al, Proc. Natl. Acad. Sci. USA, 2003, 100, 2538
12 P Hajkova et al, Nature, 2008, DOI: 10.1038/nature06714






No comments yet