Could artificial enzymes finally be about to shake up catalysis? James Mitchell Crow investigates
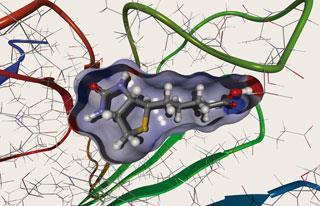
Back in 1978, a young George Whitesides made the first artificial enzyme that relied on a metal complex wedged inside a protein host.1 His proof-of-principle experiment suggested a way to improve on nature’s own catalysts with some tailor-made metal chemistry - potentially offering the best of both worlds.

But despite this early promise, wind forward to 2007 and artificial metalloenzyme catalysts still aren’t routinely used in the lab.
Now, two European chemists are finally making significant progress in the area, extending Whitesides’ original system using approaches that are - conceptually, at least - diametrically opposed. Tom Ward at the University of Neuchatel in Switzerland is tackling the problem by rational catalyst design, while Manfred Reetz at the Max-Planck Institute in Mulheim an der Ruhr, Germany, is allowing his enzymes to evolve by applying a kind of Darwinian selection. The two scientists have agreed to focus on their own particular strategies to avoid treading on each others’ toes, although the boundaries between their projects are beginning to blur.
’Everyone will tell you that the original paper was published by George Whitesides in the late 1970s, says Nick Turner, director of Bio3, the UK Centre of Excellence for biocatalysis in Manchester. ’That’s because Whitesides is well ahead of the curve. But the work that Tom Ward and Manfred Reetz are starting to publish looks very exciting.’
Best of both worlds
When choosing a catalyst, synthetic chemists are generally faced with two options: to use a metal complex, or try an enzyme as a biocatalyst. Although there has been an avalanche of recent progress in using organic molecules as catalysts (for example, see Chemistry World, May 2007, p24), chemists are still excited by artificial enzymes because they potentially combine the reactivity of traditional metal catalysts with the selectivity of enzymes.
Enzymes are made of long protein chains that wrap up to form a complex three-dimensional structure, and are particularly good at speeding up enantioselective reactions - that is, reactions in which only one of two possible mirror-image molecules, or enantiomers, form. From drugs to food flavourings, enantiopure synthetic molecules are crucial compounds.
Enzymes catalyse reactions with remarkable control by enveloping their substrate in an active site. This holds the substrate in place so that the reaction can only proceed in one orientation, selectively delivering a single enantiomer of the product. Enzymes perform so well that chemists now routinely hijack their chirality, or asymmetric shape, for their own purposes.
On the other hand, transition metal complexes have a long history of success in industrial-scale synthesis. Each metal atom can be bonded to surrounding ligands, which control the way a reactant can approach the metal. While these systems are unable to hold the reactant as precisely as in an enzyme active site, they can offer some distinct advantages. In particular, many of chemistry’s most active catalysts use the metals rhodium, palladium, platinum and ruthenium, which have never been found in enzymes. These metals can catalyse reactions that enzymes are simply unable to tackle.
In the 1970s, Whitesides - then at the Massachusetts Institute of Technology in Cambridge, US - reasoned that artificial metalloenzymes could combine the best properties of both. ’The notion was,’ says Whitesides, now at Harvard University, Cambridge, US, ’you take advantage of the chirality of a protein’s surface to influence the chirality of the environment around a rhodium atom.’
To make his artificial enzyme, Whitesides exploited the exceptionally strong binding known to exist between a small molecule called biotin and the protein avidin. By attaching the metal - in this case, rhodium - to the biotin, and then mixing it with avidin, he made a metal-protein complex. Whitesides used this system as a hydrogenation catalyst, to promote the addition of hydrogen across a carbon-carbon double bond to give a single bond.
’At the time we were doing this,’ says Whitesides, ’the options for doing the biological part were pretty limited. This was at the time when a protein crystal structure was really a big deal, and the tools for doing site-selective mutagenesis were extremely primitive. And now all that is available, what Reetz and Tom [Ward] are doing is to combine the biological approaches, the combinatorial approaches and the chemical approaches.’
Almost three decades after Whitesides’ work, scientists can now study and manipulate enzymes with some sophistication. Enzymes are made up of long chains of amino acids. There are 22 different amino acids found naturally and, by stringing these building blocks together in different sequences, nature makes proteins with a diverse range of three dimensional structures. It’s now possible to alter the DNA instructions for making a particular enzyme so that a specific amino acid in the protein chain is swapped for one of the other 21 possibilities; a process known as ’site-directed mutagenesis’. Alternatively, the amino acid can be randomly switched, scrambling the sequence at that point. Making these switches can often change the shape of the protein.
Such is the elegance of Whitesides’ biotin-avidin concept that the chemists currently exploiting these new techniques are cutting their teeth on this same hydrogenation system. The new tools developed in molecular biology in the last few years have seen Whitesides’ system finally reaching its full potential.
A European union
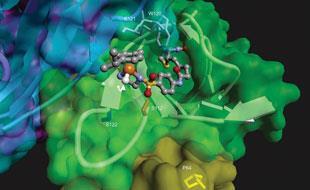
Some of the most impressive recent results have come from the team led by Tom Ward. ’I arrived at Neuchatel as an organometallic chemist,’ says Ward, ’but it soon became apparent to me that focusing on what I call the first coordination sphere around the metal was not sufficient. Enzymes work by gaining control over the whole environment. So the best way to achieve this type of control was to put our catalyst inside a protein.’
To improve the selectivity of Whitesides’ system, Ward introduced a linker between the biotin and the metal. By using linkers with different shapes and lengths, the team could place the metal centre at different positions on the protein surface. The second change the Neuch?tel group made was to switch the protein host from avidin to streptavidin, which binds to biotin with similar affinity, but inside a deeper cavity. Finally, Ward started using mutagenesis to change the amino acids which computer models suggested were close to the active site.2
Using the hydrogenation reaction to test his system, Ward screened libraries of many possible permutations of linker and enzyme mutant combinations, and discovered combinations that would give up to 94 per cent excess of one enantiomer over the other. In comparison, Whitesides’ original catalyst had achieved a modest
41 per cent enantiomeric excess (ee). Remarkably, Ward also showed that, just by changing the linker, he could go from producing 94 per cent ee of one enantiomer to 88 per cent eeof its mirror image.
The highly tunable approach meant that results came quickly. ’When we switched to streptavidin, the first experiment gave 81 per cent ee’ recalls Ward. However, working with small quantities of an air-sensitive metal caused some initial problems. ’I asked the student to reproduce it; that was carnival week so I left on vacation with my family. He repeated it three more times, and by the Wednesday of that week I returned from vacation because we couldn’t reproduce it.’ The answer was to carry out the reaction in a glove box, which excludes air. ’Ever since we bought this glove box we’ve had no problems,’ says Ward.
Survival of the fittest
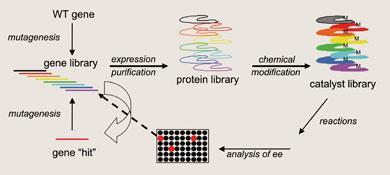
Unlike Ward, who moved into the area from organometallic catalysis, Manfred Reetz’s background is in classical enzyme catalysis. ’In the late 1990s,’ says Reetz, ’we were still using enzymes as catalysts. But people would say, "Oh yes, but enzymes can’t do this reaction or that reaction." So we decided to try these reactions using artificial enzymes, which we would engineer using directed evolution.’

Where Ward uses a rational design approach, Reetz’s technique involves generating vast numbers of artificial enzymes, and then screening for the best one. The directed evolution approach was a concept pioneered by Reetz for classical enzyme catalysis. Using random mutagenesis, Reetz would produce a large library of mutant enzymes, in the expectation of eventually forming a mutant that performed better than the parent. That improved mutant would then be used in a second round of mutagenesis. By successively selecting the best catalyst from each generation of mutants, this was a case of survival of the fittest catalyst. The key to this approach is being able to screen huge libraries of mutants to find the best one. While working on classical enzyme biocatalysts, Reetz was typically producing 3000 mutants per round of mutagenesis, of which 500-800 could be screened for activity each day, thanks to automation and high-throughput analysis. However, converting each mutant into an artificial enzyme and then screening it is a far trickier proposition than screening classical enzyme biocatalysts.3 As Ward had found, the transition metal can often be quite air-sensitive. Using Reetz’s approach, which relies on producing and screening huge numbers of analogues, glove box conditions are hardly an option.
So Reetz’s team is currently restricted to making and screening a few hundred mutants per generation of catalyst, with limited automation. ’But a dog with only three legs can still run,’ laughs Reetz. ’So we use the system to demonstrate proof of principle, that over several evolutionary cycles we produced a better catalyst.’4 Reetz is still working towards an automated, high throughput approach to make and screen large artificial enzyme libraries. ’Hopefully within the next two years,’ says Reetz, ’we or another group will find a new protein scaffold that is stable, has a high expression rate, and is easy to purify. Then we can start working on reactions that are currently difficult to do, such as ring closing metathesis, Grubbs chemistry or regioselective hydroformylation.’
Meanwhile, other researchers are already exploring the potential of wrapping metal complexes up in DNA, rather than proteins.
Into the future
’I think the potential - and I know this is what Tom and Manfred are looking at - is to try to generate really novel activities,’ says Nick Turner. ’The best thing is not to go after activities that are already pretty established using conventional enzymes, but really to try to use the transition metal centre to introduce a new type of chemistry that is not currently possible using existing enzymes.’
And once these new reactions are developed, could they be used industrially? ’There’s no reason why they shouldn’t for some cases,’ says Whitesides, ’because enzymes themselves are increasingly used in process chemistry. And these systems could be cheap.’ In fact, Ward is already taking steps in this direction. ’We have one hydrogenation substrate that we’re collaborating [on] with industry, where there is no homogeneous catalyst capable of performing that reaction,’ he says.
However, Reetz reckons that the potential for artificial enzymes in industry is ’difficult to predict. If I was an assistant professor in the US, I would say, "of course!" But having been in research for 35 years, I feel I must be optimistic but realistic.’
So which strategy is likely to win out - the rational approach of Ward, or Reetz’s directed evolution?
’It’s a mixture of both, it’s whatever’s going to get you to the end point,’ says Turner. ’The way the field is going, we take any piece of information we can get hold of, and feed it into the algorithm,’
Ironically, Ward is one of the greatest supporters of Reetz’s approach. ’I think there are technical hurdles to be overcome with the directed evolution, but I think these can be overcome, and then I think that will be the most straightforward way to do it,’ he says.
Whitesides thinks that the implications of this research reach far wider than catalysis. ’We’re very interested in the area of rational drug design, which in some senses is exactly the same problem; that is, how do you design a ligand in such a fashion that, when you know the structure of the surface of a protein, it will stick well?’
’When I first entered chemistry, it was important for chemistry to understand the design rules for making ligands that bind to the active sites of enzymes,’ he says. ’Now we’re going on 50 years later and this is still a very important problem. That, to me, is the ultimate justification for this - the real home run in this area is to understand molecular recognition.’
References
et al, Chem. Commun., 2006, 4318
5 G Roelfes, A J Boersma and B L Feringa, Chem. Commun., 2006, 635












No comments yet