

Chiral control through halogen bonding could be the next frontier for organocatalysis
A long-overlooked mode of catalysis is finally going asymmetric. Halogen bonds were first proposed as an activating interaction in 2008, but this alternative type of organocatalysis was initially slow to gain traction among the organic community. General scepticism about their paradoxical nature, coupled with the difficulty of engineering an effective chiral catalyst, led many to dismiss halogen-bond donors in favour of more established hydrogen bonding systems.
However, halogen-bond catalysis has quietly been undergoing a revolution. Deepening insights into this unusual non-covalent bond over the last 15 years have boosted the performance of many halogen-bond systems well beyond the capabilities of conventional organocatalysts. And now, a flurry of asymmetric approaches are broadening the reach of halogen-bond catalysis even further, slowly bringing it to the mainstream as an alternative tool in organic synthesis.
‘I personally think the reason these bonds have been underappreciated is the Lewis structure,’ says Stefan Huber, an organocatalysis researcher at Ruhr University Bochum in Germany. ‘It’s a very counterintuitive interaction and there are many different contributions.’
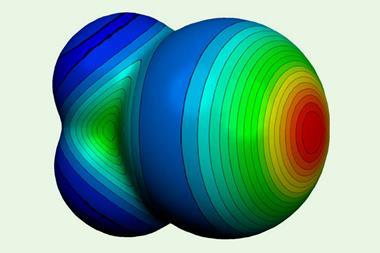
Halogen bonds are a weak intermolecular interaction between a localised electropositive region on a halogen atom and a nucleophilic region on another molecule. Poor orbital hybridisation in the heavier halogens creates a small gap in the electron density surrounding the atom, opening up an area of positive charge from the core known as a σ hole. Electrostatic attraction between this localised positive area and a nearby nucleophilic atom forms a weak intermolecular bond that withdraws electron density from this bound substrate. At the same time, the σ* antibonding orbital in the halogen-bond donor is polarised towards the halogen, creating a low-lying, attractive orbital with a huge lobe on the halogen that is easily filled by a Lewis base.
In principle, these bonds activate substrates in much the same way as hydrogen bonding; weakening a bond in an electrophile to ready it for attack by a nucleophile. Like other organocatalysts, many halogen-bond donors are also relatively user-friendly, demonstrating good tolerance towards air and moisture, and a reduced environmental footprint compared with metal catalysts. But, while hydrogen-bond catalysis flourished in the 1990s, these analogous non-covalent interactions were largely ignored.
What is halogen-bonding catalysis?
A quick introduction to halogen bonds and how chemists are using them
What is a halogen bond?
A halogen bond is a non-covalent interaction between a covalently bonded halogen atom and a nucleophile.

The attraction is often explained by invoking the σ-hole concept – a region of low electron density that arises on the halogen (X) when it is bound to an electron-withdrawing group (EWG). However, charge transfer from the nucleophile into the σ* antibonding orbital can also play a role.

Halogen bonds are comparable in strength to hydrogen bonds, but they are much more directional.
How are they used in catalysis?
Just as chemists harness hydrogen bonds when designing organocatalysts, researchers have started using halogen bonds to promote a variety of reactions.
A common strategy uses bidentate halogen-bond donors. In this Diels–Alder reaction, the catalyst enables the reaction to proceed quantitatively at room temperature, whereas no product forms in the absence of the catalyst.

The halogen bonds between the catalyst's iodine atoms and the carbonyl oxygen activate the carbonyl carbon to nucleophilic attack.
What about enantioselective catalysis?
Using halogen bonds in asymmetric catalysis is challenging because of their directionality and the long distance between the substrate and the catalyst framework.
The first enantioselective halogen-bonding catalyst was reported in 2020. This achieved moderate enantioselectivity in a Mukaiyama aldol reaction using a bidentate halogen-bond donor to impose a chiral environment around the substrate.

Chemists hope that chiral halogen-bond catalysis can be added to the organocatalytic toolkit for situations where hydrogen bonding doesn’t work.
Halogen-bond catalysis begins
This all changed in 2008 when Carsten Bolm postulated halogen bonding as the mode of catalytic activation for a simple quinoline reduction.1 He proposed that a halogen–nitrogen interaction between a haloperfluoroalkane catalyst and quinoline substrate activated the latter for reduction by a Hantzsch ester, giving the tetrahydro product in 98% yield.

While this was the first suggestion of catalytic activation via halogen bonding, today, the true mode of catalysis in Bolm’s reaction is still somewhat disputed. ‘The biggest enemy in proof-of-principle studies is protons,’ explains Huber. ‘So often you have acid traces somewhere, which could also catalyse these reactions. To rigorously conclude it’s halogen-bond catalysis, you have to exclude acids and all other mechanisms.’
Despite these mechanistic doubts, the report sparked an idea for Huber and, inspired by Eric Jacobsen’s thiourea catalysts, he developed a bidentate cationic halogen-bond donor for a straightforward solvolysis reaction.2 Crucially, the team took pains to demonstrate that the analogous hydrogen compound was inactive, ruling out hydrogen bonding and providing incontrovertible evidence of halogen bonding as the mode of activation.
‘These cationic systems were the first that really worked, but there are three types of halogen-bond donors: neutral backbone, cationic backbone and hypervalent. Which you use depends on your substrate and reaction,’ says Huber.
Bolm’s perfluoroalkanes are an example of a neutral donor, which typically use a fluorinated backbone to withdraw electron density from the activating carbon–iodine bond. Relatively stable and readily soluble in organic solvents, these are the weakest class of halogen-bond donor catalysts and are insufficiently Lewis acidic to activate carbonyl compounds. Cationic systems exert a stronger pull on the substrate and are essentially the halogen equivalent of charge-assisted hydrogen bonds, making them substantially more active than their neutral counterparts and comparable or better than equivalent hydrogen-bond catalysts. Meanwhile, hypervalent iodine catalysts are the most Lewis acidic. Although powerfully activating, these systems are vulnerable to decomposition and careful catalyst design is required to stabilise the iodine centre.
Broadly speaking, halogen-bond donors catalyse the same types of reactions as hydrogen-bond catalysts including, Mannich reactions, Michael additions, halide abstractions and Diels–Alder reactions. ‘In the first five or six years, we used very simple benchmark reactions just to demonstrate that it’s possible,’ says Huber. ‘Clearly that’s not something that’s interesting from a synthetic perspective but since the very first paper, everybody said “can you do that enantioselectively?”.’
In pursuit of enantioselectivity
Several innate characteristics of the halogen bond interaction make this an extremely challenging endeavour. The carbon–iodine bond is around 3Å long, meaning that the point of activation is generally a long way from the chiral centre on the catalyst. This, coupled with the high directionality of the halogen bond itself – 180° from the carbon–iodine bond in the catalyst – means transferring the stereochemical information therefore requires an extensive bulky structure to encapsulate the substrate. Such highly-designed catalysts are not commercially available and effective structures often require lengthy syntheses to build up the necessary complexity.
‘Our first approach was to use known halogen-bond donor systems and attach chiral sidearms in a single bond, but we saw no enantioselectivity at all,’ recalls Huber. After several unsuccessful iterations, the team proposed that free rotation of the sidearm may cause an averaging effect, cancelling out any selectivity over the course of the reaction to give racemic products. This initial failure highlighted the second essential requirement of an effective chiral catalyst – structural rigidity.
Finally in 2020, the group published the first example of enantioselectivity, albeit moderate, in halogen-bond catalysis. Using a similar bidentate cationic catalyst, this time with two chiral side arms each tethered by two bonds, they created a rigid chiral pocket and, in a proof-of-concept demonstration, performed an asymmetric Mukaiyama aldol reaction with 30% enantiomeric excess.3
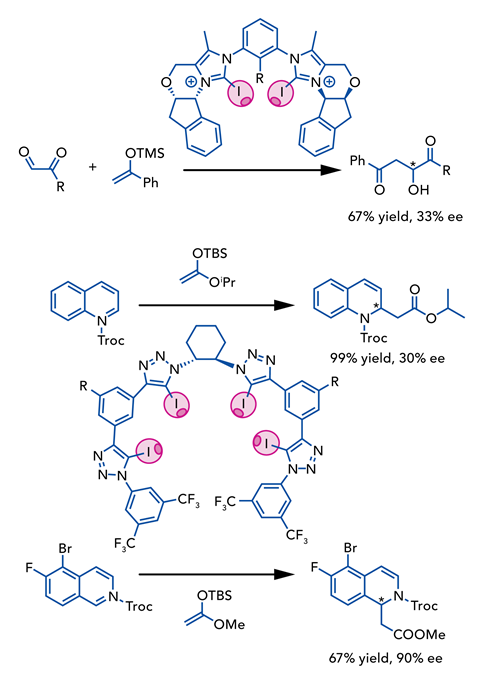
This was quickly followed by a neutral example, developed by Olga Garcia Mancheño at the University of Münster in Germany. Her team used a tetrakis-iodo-triazole – a bulky tetradentate unit containing four iodine centres positioned within a stiff chiral pocket – to activate a carbon–chlorine bond for a Reissert-type dearomatisation of quinoline, which likewise gave a moderate enantiomeric excess of 30%.4 Further investigation revealed that by incorporating a carbon–halogen bond into the substrate, they could introduce an additional halogen bond between the substrate and catalyst, boosting the enantiomeric excess to over 90% for these engineered substrates.5
A combined approach
However, the first example of a highly enantioselective halogen-bond catalyst was reported by Yasushi Yoshida at Chiba University in Japan in 2021.6 Rather than relying exclusively on halogen bonds to activate and position the substrate, the team designed a bifunctional catalyst system, incorporating both halogen and hydrogen bond moieties in a binaphthyl skeleton.
Unusually, Yoshida chose bromine, not iodine, as the halogen-bond donor. ‘Iodine can make a stronger halogen bond than bromine or chlorine, but our catalyst design is bifunctional, with hydrogen bonding from the benzyl amide,’ he explains. ‘The balance of the halogen bonding with the hydrogen bonding is important for making a good asymmetric environment around the prochiral carbon and the reaction occurs in a pseudo-intramolecular fashion to give an enantioselectivity increase.’
The team’s system is a rare example of hypervalent bromine acting as catalyst, and Yoshida’s bromonium salts facilitated a vinylogous Mannich reaction of cyanomethyl coumarins with ketimines in up to 96% enantiomeric excess. This was quickly followed by an analogous iodine system, which they demonstrated in the asymmetric synthesis of N,S acetals.7
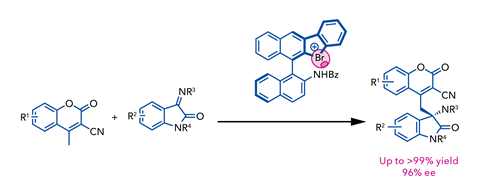
Designing these hypervalent catalysts, however, is challenging. Lengthy synthetic sequences limit their application to the small subset of chemists intentionally studying these interactions. Meanwhile, the active iodonium and bromonium salts are both fairly unstable, particularly towards light, heat and acid, making catalyst synthesis and purification difficult. Proving the role of any halogen-bond interactions in the activating mechanism is therefore a key part of reaction development. ‘We tried the reaction with a chiral binaphthyl-based catalyst with only the benzyl amide moiety and in that case, there was almost no enantioselectivity. It’s not direct evidence, but I believe the halonium salt moiety and the halogen bond are important for both the selectivity and activity in that reaction,’ says Yoshida.
Stripping it back
The sheer complexity of current halogen-bond catalysts is one of the big limitations preventing the field from becoming more established, says Boris Nachtsheim, an organo-iodine chemist at the University of Bremen in Germany. ‘The catalysts out there are either simple but not high-performing or they are very complex. The trade-off for selectivity is synthetic complexity,’ he says. ‘So I wanted to make a catalyst that is easy to modulate on one hand, but on the other hand also has enough enantio-induction.’
Rather than engineering a completely new system, Nachtsheim merged two existing research lines within his group, combining N-heterocyclic hypervalent iodine compounds – originally developed for oxidative transformations – with diaryliodonium salts used for the synthesis of heterocycles. The result was a chiral triazole-substituted iodonium salt, featuring a hypervalent iodine centre with a tethered chiral sidearm.8
Crucially, despite monodentate binding with the substrate, additional coulombic and orbital interactions hold the entire system rigid, preventing the free rotation that erodes selectivity. The team screened the modular system against several addition reactions including a vinylogous Mannich and N,O-acetal formation, obtaining enantiomeric excesses of greater than 98%.
But while the catalyst structure and synthesis are relatively straightforward, the mechanism of enantio-induction involves a complex sequence of interactions. ‘A coulomb interaction of the iodonium salt with the triazole holds the chiral sidearm in place and takes away the rotational degrees of freedom,’ explains Nachtsheim. ‘Then there are two halogen-bond interactions, an intermolecular one with the carbonyl of the substrate carbamate, and an intramolecular one with the benzyl ether of the catalyst. Both substrates (catalyst and carbamate) also coordinate via CH–π and π–π interactions, forming a stiff iodonium-centred conglomerate.’
Switching up the mechanism
Exploiting interactions between other parts of the system was also key to Huber’s latest chiral strategy, developed in collaboration with chemistry Nobel laureate Benjamin List. ‘Instead of using a chiral donor with a “boring” counterion, we use a simple halogen-bond donor with a chiral counterion,’ he says.
List and Huber combined an achiral bidentate halogen-bond donor di-cation with two equivalents of a chiral disulfonimide counterion to form a highly ordered super-aggregate.9 The resulting structure has a central binding position, enabling a carbonyl substrate to form a halogen bond to each of the iodonium centres, while the counterions create a chiral pocket, likewise associating with the halogen-bond donor cation via peripheral halogen bonds. ‘There’s a sweet spot in the choice of counterion,’ Huber explains. ‘If it’s too coordinating, like a phosphate, it blocks your iodonium. If it’s almost non-coordinating, like the imidodiphosphorimidates, it doesn’t form this nice pocket. The disulfonimide was just right and works with high enantioselectivity – up to 96% enantiomeric excess – in a relatively challenging Diels–Alder reaction.’

A tool for synthesis?
While halogen-bond catalysis has made huge progress in the last five years, broadening the appeal and ease of access of these systems will be key to making this chemistry a more general synthetic tool. The majority of the work since Bolm’s seminal paper in 2008 has been to establish the basics of reactivity and selectivity. For Huber, the natural next step is for halogen bonding to find its own particular mechanistic niche.
‘I have this vision that 20 years down the road, we will have this map in the field of non-covalent catalysis where some substrates go towards hydrogen bonding and some substrates go towards halogen bonding,’ he says. ‘It’s a matter now of further developing this, having broader databases of what works, and making halogen-bond catalysis more attractive to people who buy catalysts from a catalogue.’
References
1 A Bruckmann et al, Synlett., 2008, 6, 900 (DOI: 10.1055/s-2008-1042935)
2 SM Walter et al, Angew. Chem., Int. Ed., 2011, 50, 7187 (DOI: 10.1002/anie.201101672)
3 RL Sutar et al, Angew. Chem., Int. Ed., 2020, 59, 6806 (DOI: 10.1002/anie.201915931)
4 F Ostler et al, Chem. Eur. J., 2021, 27, 2315 (DOI: 10.1002/chem.202005016)
5 AC Keuper et al, Angew. Chem., Int. Ed., 2023, 62, e202304781 (DOI: 10.1002/anie.202304781)
6 Y Yoshida Yasushi, T Mino and M Sakamoto, ACS Catal., 2021, 11, 13028 (DOI: 10.1021/acscatal.1c04070)
7 Y Yoshida et al, Adv. Synth. Catal., 2021, 364, 1091 (DOI: 10.1002/adsc.202101380)
8 M Damrath et al, ACS Catal., 2025, 15, 422 (DOI: 10.1021/acscatal.4c06895)
9 DL Reinhard et al, J. Am. Chem. Soc., 2025, 147, 8107 (DOI: 10.1021/jacs.4c18378)












No comments yet