
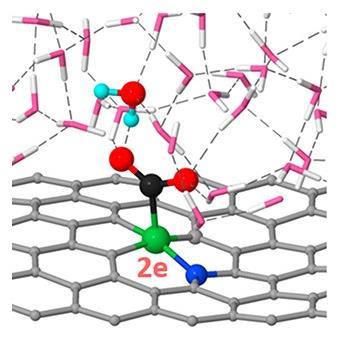
Hydrogen bonds and charge capacity are the secret to nickel atoms’ outstanding ability to reduce carbon dioxide, researchers have discovered. Understanding the reaction mechanism better could lead to more efficient catalysts for turning carbon dioxide into useful chemicals – like fuels or materials – to reduce our dependence on the fossil fuels that generate the gas fuelling climate change in the first place.
Using electrochemistry to reduce carbon dioxide into carbon monoxide is tricky, as the reaction’s potential is very similar to that for hydrogen production. ‘Single-atom nickel on carbon is a great solution – it’s an abundant metal on a cheap conductive support and offers great atom efficiency,’ explains Núria López, a theoretical chemist at the Institute of Chemical Research of Catalonia, Spain, who wasn’t involved in the study. But so far chemists have failed to explain why nickel is so good at favouring carbon dioxide conversion over the competing reaction.
‘Previous computational calculations were overlooking some very important factors,’ says the study’s leader Yuanyue Liu from the University of Texas at Austin, US. ‘Our approach considers aspects like hydrogen bonding and surface net charge, key to the electrochemical reaction.’
In ab initio molecular dynamics, Liu’s team discovered that some of the intermediates in the conversion of carbon dioxide to carbon monoxide are highly polar and interact strongly with the water molecules around them. ‘These strong interactions – both hydrogen bonds and strong charge effects – could explain the high selectivity of nickel catalysis towards the reduction of carbon dioxide,’ says Liu. The intermediates of the hydrogen evolution reaction tend to be neutral species – they don’t form hydrogen bonds with water. ‘Surface charge is key to the activity,’ remarks Liu. ‘If the site has a higher charge capacity, it can carry more net electronic charge, accelerating the electrochemical reaction.’
López thinks this study offers an interesting solution beyond the classic density functional theory methods. ‘Computational chemists have been using the same techniques for over 10 years, and spicing that up with molecular dynamics is the next step needed,’ she says.
Molecular dynamics calculations represent the system better, but require ‘orders of magnitude more computing power’, López adds. She explains that previous approaches used ‘ad-hoc corrections’ that only estimate the effect of charge interactions, including hydrogen bonds. ‘Hydrogen bonds are too dynamic, simulating that with a computer is complicated,’ explains Liu.
However, López says these results need to be corroborated by experiments in the lab. Further studies will have to compare the results to spectroscopic fingerprints and identify the real active sites. ‘Active site speciation is the key of single-atom catalysis,’ she says. ‘It’s an interesting proof of concept, but additional experiments need to prove the suggested active species is stable under the calculated electrochemical conditions.’
References
X Zhao and Y Liu, J. Am. Chem. Soc., 2020, 142, 5773 (DOI: 10.1021/jacs.9b13872)












No comments yet