Ability to map phase diagrams using quantum calculations could benefit pharma research
Envisaging the laboratory conditions under which desired polymorphs will form is now on the horizon thanks to new computational techniques developed by scientists in the US. Using first-principles quantum chemistry, the scientists have been able to derive the polymorph phase diagram of methanol to a level of accuracy that holds great promise for pharmaceutical applications.
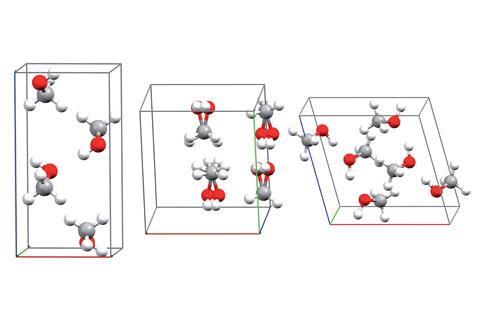
A given molecule can pack together in many different ways when it forms an ordered solid arrangement as a crystal structure. Different crystal structures of the same molecule are known as polymorphs. Despite being formed of identical components, polymorphs can have vastly different physically properties. This is relevant to the pharmaceutical industry, where two polymorphs of a drug molecule may interact with the body in dramatically different ways. Searching for polymorphs experimentally by trial-and-error is laborious; scientists have therefore sought to streamline the process. This led a field known as crystal structure prediction, where scientists attempt to predict a compound’s crystal structure based only on its composition. However, these predictions frequently ignore real-world conditions, such as temperature and pressure, limiting their accuracy.
Greg Beran, from the University of California, wanted to make these predictions more valuable to those working in the lab: ‘What we thought would be really useful is not only to be able to say what crystal forms might occur, but be able to tell experimentalists under what conditions these would occur.’ Beran’s group has now used fragment-based quantum chemistry to study a molecule and its immediate neighbours separately and led them to derive a phase-diagram showing the different temperatures and pressures for which the polymorphs of an organic molecule can form, with much more accuracy than previous crystal structure prediction methods would have allowed.
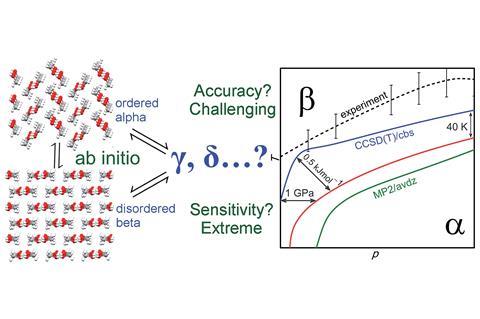
The group chose methanol to test their calculations. ‘Methanol actually possesses many of the difficulties of larger molecules,’ explains Beran, such as many long-range and weak interactions, and required a few hundred thousand computing hours to model. Nonetheless, by the end the group accurately predicted the phase diagram of methanol to within 0.5kJ/mol, a result that puts this work amongst the most sophisticated predictive models for polymorphs.
Graeme Day from the University of Southampton, UK, an expert in crystal structure prediction, is impressed with the research. ‘To map out the whole temperature–pressure phase diagram for a molecular crystal and to get it even qualitatively correct is a big achievement,’ he states.
‘Finding more complex molecules where you can validate the method might get tricky, but I hope they are going to try out these methods on real pharmaceutical-type molecules and see how they do. That would be very exciting and really guide those people into looking for structures or pinning down experimental conditions under which you can get polymorphs,’ adds Day. Beran agrees that getting to the point of predicting phase diagrams for small pharmaceutical molecules is when the work will have a real impact.
References
This article is open access
C Červinka and G J O Beran, Chem. Sci., 2018, DOI: 10.1039/c8sc01237g









1 Reader's comment