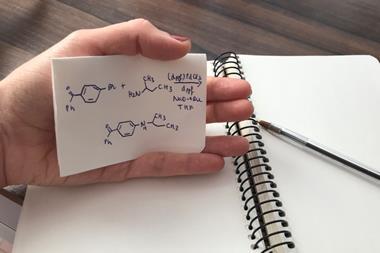
A system involving a cationic iridium catalyst has enabled the first enantioselective addition of an N–H bond to an unactivated internal olefin. The new strategy provides a starting point to overcome a long-standing challenge in organic chemistry and is an important step towards functionalising basic feedstocks for different uses. ‘We revealed an approach to add N–H bonds to carbon–carbon double bonds within a set of alkenes that have rarely undergone this reaction before and never with control of the absolute configuration of chiral products,’ says John Hartwig at the University of California, Berkeley in the US, who led the study.
Chiral amines are key structural motifs in many drugs and agrochemicals, so finding efficient ways to prepare these compounds is an important goal in chemical synthesis. Kai Hultzsch at the University of Vienna in Austria, who was not involved in the study, explains that traditional amine syntheses usually involve many reaction steps and use refined compounds as the starting materials. ‘The hydroamination reaction, in which an amine N–H is added across an unsaturated carbon–carbon bond, uses simple and inexpensive olefins directly from petrochemical feedstock and – ideally – ammonia as the nitrogen source,’ he says. But Hultzsch also stresses that a catalyst is required and despite all the efforts made over the last three decades to improve this desirable reaction, it remains challenging.
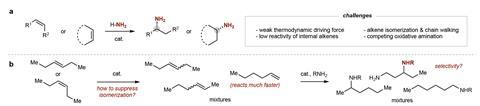
Matthew Gaunt at the University of Cambridge in the UK adds that alkenes in which the carbon–carbon double bond is in the middle of the alkyl chain are particularly unreactive. ‘Moreover, in cases where the alkene has two different substituents, controlling which end of the carbon–carbon double bond reacts with the nitrogen atom of the amine is difficult.’
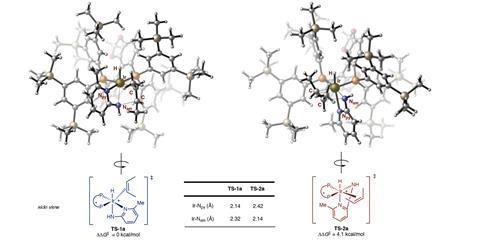
Using a special design, the US team was able to overcome these limitations. ‘Our system comprises an alkene and an amine that react in the presence of a soluble transition-metal catalyst based on iridium that is cationic and contains a new phosphine ligand,’ Hartwig says. He explains that cationic iridium catalysts are more active than neutral ones in this type of reaction, ‘presumably because they bind the alkene with greater equilibrium constants and because cationic complexes tend to undergo migratory insertion faster than analogous neutral ones’. Hartwig points out that the reaction benefits from three components. ‘First, the amine contains a pyridyl group that is designed to bind weakly to the metal but assists cleavage of the N–H bond. Second, the ligand contains trimethylsilyl groups in the periphery – the origin of the beneficial effect of those groups is unclear. Third, the triflimide counteranion strongly influences the overall rates, as well as relative rates for addition versus olefin isomerisation,’ he says. DFT calculations helped the scientists to understand the reaction mechanism and uncover the possible origins of enantioselectivity.
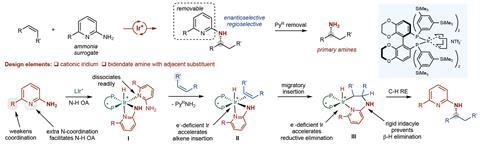
‘Obviously the phosphine ligand is controlling a lot of the selectivities in this reaction,’ comments Gaunt. ‘But the amine itself is also quite cleverly designed. It needs to coordinate to the iridium and allow the N–H bond to be activated, but also not to coordinate too strongly so that it deactivates the catalyst.’ Hultzsch adds that the pyridine moiety can be cleaved after the hydroamination reaction to obtain a primary amine. ‘The obtained product constitutes the hydroamination product as if ammonia had been used,’ he notes. ‘While the removal of the pyridine moiety reduces the overall atom efficiency of the process, a direct stereoselective hydroamination using ammonia is not known so far.’
The researchers believe their results could also open the way to advances in other alkene functionalisation reactions including the addition of N–H bonds from other compounds besides amines or the addition of O–H bonds from alcohols or carboxylic acids.
References
Y Xi, S Ma and J F Hartwig, Nature, 2020, DOI: 10.1038/s41586-020-2919-z










No comments yet