Porous materials made from small molecular cages, rather than rigidly bonded frameworks, could be easier to process and have more tunable performance, say UK researchers. The team is also developing models to predict the way the molecules pack together, which is crucial in determining their physical properties.
‘Zeolites and metal-organic frameworks (MOFS) don’t really dissolve,’ says Andrew Cooper from the University of Liverpool, UK, speaking at the 10th International Symposium on Advancing the Chemical Sciences in Kyoto, Japan. He explains that the smaller molecules that make up his team’s materials are soluble, which means they can be processed into a variety of shapes, such as films or fibres. That could be an advantage when it comes to preparing the materials for their eventual applications.
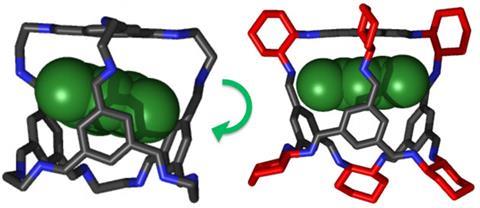
Porosity without pores
One feature of molecular crystals is that they are less rigid than their MOF cousins. ‘There’s molecular vibration all the time, which means the windows into the cages are opening and closing slightly. That means molecules that look too large can fit through the pores,’ Cooper explains. This can make predicting absorption selectivity of guest molecules from just the crystal structure quite treacherous, but opens up the possibility of harnessing that fluctuation to reversibly catch and release molecules under different conditions.
It’s this unpredictability that has held back crystal engineering for molecular solids, says Cooper. ‘What we’re trying to do is develop ways to control, or at least predict, the assembly,’ he adds. Together with Graeme Day at the University of Southampton, UK, the group has developed a model for predicting the cages’ crystal packing.
Cooper suggests that the same kind of models could be applied more widely to try and work out some basic rules for how all sorts of molecules pack into crystalline solids. ‘You don’t need to feed in experimental data to get the calculations to work,’ Cooper says. ‘All you feed in is the molecular formula, so it can work for hypothetical molecules you haven’t made yet.’
While the calculations are still quite computationally expensive at this stage, spending two days on calculations is still more efficient than several weeks in the synthetic lab to create new cage designs, which then might not even crystallise the way you want them to.
Susumu Kitagawa, who researches porous materials at the University of Kyoto, Japan, emphasises the importance of combining theory with experiments to gain predictive power. Many people have made similar cage compounds, he observes, but predicting their structures and properties is difficult. ‘This work is opening up a new field, where well-ordered assembly can lead to materials with beautiful new properties,’ he says.






No comments yet