A new high-throughput technique can analyse the folding stabilities of nearly one million protein sequences at a time. The unprecedented method – which is fast, accurate and scalable – promises to help understand how amino acid sequences fold into three-dimensional conformations, while providing the data needed to improve machine learning models.
The propensity for proteins to fold spontaneously is governed by their hidden and subtle energetics – such as hydrogen bonding and hydrophobic effects – that are unique to any given amino acid sequence that makes up a protein. Since even a single mutation in a protein’s sequence can affect folding, measuring stability is important for understanding disease, as well as drug development and protein design.
However, for decades it has only been possible to measure protein folding stability in a few proteins at a time. While thousands of measurements have been gathered, there’s still not enough data for machine learning to begin predicting and unravelling the hidden thermodynamics of folding stability.
Now, an international team of researchers, headed by Gabriel Rocklin’s lab at Northwestern University Chicago, US, has developed a method that can measure protein folding stability in parallel for as many as 900,000 protein sequences up to 72 amino acids long. The researchers measured stability for 1.8 million sequences in total and filtered their data to obtain 776,000 high-quality folding stabilities.
‘Predicting the stability of an arbitrary protein sequence has always been an impossible dream – proteins were viewed as just too complex,’ says Rocklin. ‘But with millions of data points from this technique, I believe we’ll be able to develop accurate [machine learning] models. This will illuminate a huge amount of biology and accelerate the design of new, more sophisticated protein structures.’
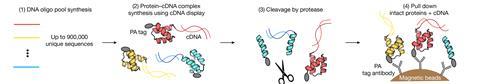
The new technique relies on cDNA display – a screening method first developed in 2009 that links proteins to DNA. Using this, the team first produced a mixture of proteins from the library of 900,000 sequences, with each protein attached to the DNA that encodes it. Then, using a decades-old method called proteolysis, proteins were incubated with proteases that degrade unfolded proteins faster than folded ones. The non-degraded proteins were then isolated and their attached DNA was sequenced to find out what they were.
‘By doing this at many different enzyme concentrations, we can figure out precisely how strongly folded each different protein is,’ explains Rocklin. ‘Lastly, we added some correction terms to relate this stability against degradation to thermodynamic folding stability.’
‘This is potentially valuable stuff,’ comments emeritus professor Alan Cooper, an expert in protein folding and thermodynamics at the University of Glasgow, UK. ‘It is an impressive way to screen a vast array of protein mutants for protease susceptibility, possibly related to folding stability.’ However, he has doubts whether protease susceptibility is a sufficient measure of thermodynamic folding stability based on inferred ratios of folded to unfolded species.
Nevertheless, Rocklin says that the large dataset they produced is already proving useful for many labs trying to develop machine learning models to predict protein folding stability. ‘Along with designing new proteins, we also want to understand how variants in our genomes influence protein folding stability that can often cause disease,’ he says. ‘Today we have a limited understanding of most genetic variants. Improved computational models can make a big impact by predicting the stability effects of these variants,’ Rocklin adds.
Correction: The 4th paragraph was updated on 28 July 2023 to correct several technical details
References
K Tsuboyama et al, Nature, 2023, DOI: 10.1038/s41586-023-06328-6










No comments yet