A new technique that can make libraries of cyclic peptides will enable researchers to probe their therapeutic benefits
Japanese researchers have developed a way of reprogramming the genetic code and using bacteria to make and screen huge libraries of cyclic peptides using unnatural amino acids. This technique could be used to develop new kinds of drugs and biochemical probe molecules.
Speaking at the RSC’s 5th International Symposium on Advancing the Chemical Sciences in Manchester, UK, Hiroaki Suga from the University of Tokyo outlined how the technique, which he calls Random Peptide Integrated Discovery, or Rapid, works and his hopes for it.
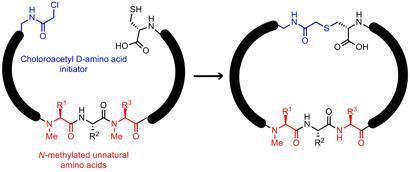
Cyclic peptides like the naturally occurring immunosuppressant drug cyclosporin A are an important class of potentially useful medicinal compounds, Suga explains. But working out the structural features that lead to therapeutic activity is difficult. ’We know that the extra rigidity of cyclic peptides compared to linear ones is important,’ Suga says, adding that incorporating unnatural N-methylated or right-handed D-amino acids - rather than the usual L-form - helps them to resist being broken down by the body’s protective protease enzymes.
Many of these compounds, which are rings that tend to be made of between five and 15 amino acids, cannot cross cell membranes, making them useless as drugs. In order to screen large numbers of them, Suga’s team developed a way of hijacking the cells’ protein making machinery (the ribosome) to produce libraries of cyclic peptides with trillions of permutations.
The system is based on protein synthesis. Normally, a short sequence of RNA is fed through the ribosome, with each set of three bases on the RNA coding for an amino acid. Those amino acids are delivered to the growing peptide chain by transfer RNAs (tRNAs) that match up with the coding strand. To include unnatural amino acids in their molecules, the team had to reprogramme the genetic code.
They attached unnatural amino acids to standard tRNAs in place of the normal amino acids that those tRNAs would code for. By mixing together this new set of tRNAs with libraries of random RNA coding sequences and the ribosome machinery (in test tubes rather than living cells), they could create libraries of eight to 11 amino acid strings, including some of the unnatural methylated amino acids. Each string was programmed to start with a chlorinated D-amino acid and end with a sulfur-bearing cysteine, which would react together to close the ring. ’Incorporating D-amino acids along the chain is difficult,’ says Suga, ’but it’s OK to begin with.’
Philipp Holliger, who researches RNA-based catalysis and replication at the Laboratory of Molecular Biology in Cambridge, UK, is impressed by Suga’s work. ’These compounds have huge potential to be useful for biotechnology and as a platform to discover new drugs,’ he says. The simplicity and scope of the system is also appealing, he adds, ’making that many molecules by chemical synthesis alone would be impossible’.
To harness these libraries for applications, Suga’s team simply introduce magnetic beads labelled with a target they want to bind - say an enzyme or drug receptor. The strongest binding peptides can then be fished out, sequenced and chemically synthesised to further check their properties. By screening such large numbers, he hopes to work out some patterns that confer useful properties like membrane diffusion. ’This method allows us to combine the power of biology and chemistry we use biology to find the best molecules, then chemistry to make them in large quantities,’ he says.
Suga has patented the Rapid process and spun out a company called PeptiDream to commercialise the technology.
Phillip Broadwith






No comments yet