An algorithm for performing challenging multistep retrosynthesis and predicting the most efficient synthetic pathway has been extended to libraries of compounds with different substitutions or isotope labels. This broadens the utility of the computer program Chematica, which interweaves mechanistic rules, quantum calculations and artificial intelligence to predict the best synthesic routes.
Retrosynthetic analysis can be a challenge for many chemists – not just undergraduates. Those that are comfortable with the retrosynthetic analysis of a single compound may still find it difficult to propose a route that efficiently creates a selection of similar molecules with differing R groups. ‘Chemists are not trained to optimise global plans,’ says Bartosz Grzybowski at the Polish Academy of Sciences, who led the software development.
‘Even when an expedient synthesis for a target compound exists, it may fail to deliver structural analogues due to functional group incompatibility, access to starting materials or cost,’ comments Robert Paton, a computational chemist at Colorado State University, US, who was not involved in the work. And Olaf Wiest, director of the Americann National Science Foundation Centre for Computer Assisted Synthesis, notes that distinct advantages arise when programs such as Chematica provide new insights into potentially more efficient pathways. ‘Optimising several objectives, such as yield, cost and step count, across complex synthetic campaigns is one area where computational synthesis planning can be expected to augment human expertise.’
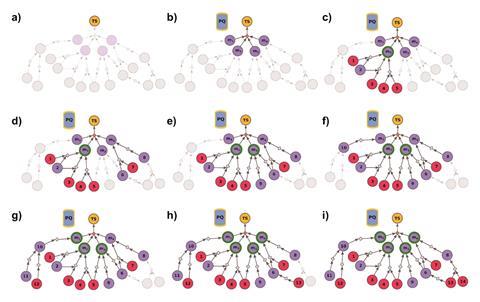
By combining expert chemical knowledge with repeated searches, Chematica can plan the synthesis of pre-selected molecules. Chematica is also able to suggest entirely new routes to compounds, avoiding patented methods or offering more efficient, cheaper and accessible synthetic routes compared with those reported in the literature.
While Grzybowski’s team has validated Chematica for the retrosynthetic analysis of single molecules, computationally designing routes to a selection of structures that differ only slightly has proven challenging. This includes pharmacologically relevant problems such as synthesising a library of compounds for exploring how slight changes alter the efficacy of potential drugs or designing molecules with different isotope labels for metabolic studies.
Going down the same path
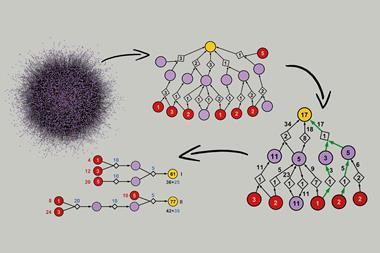
Now, a computational trick has allowed the team to expand their algorithm to work on libraries of compounds. They updated Chematica so that at the very start of the search for viable synthetic routes ‘there is a fictitious multicomponent reaction, one step that doesn’t make any chemical sense but is just for the computer to do the right thing,’ explains Grzybowski. This fictitious multicomponent reaction occurs between all members of the library under investigation and relates these members to a generalised structure.
Not only does this ensure that Chematica considers all members of the library when it hunts down the most viable synthetic routes, but also results in pathways containing common intermediates. This speeds up the computational search and results in an easier time for synthetic chemists.
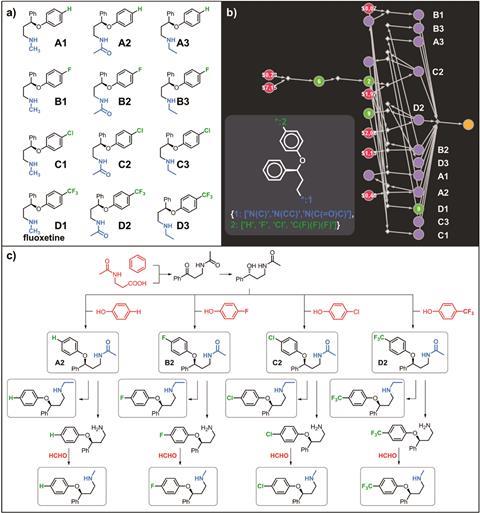
By traversing through common intermediates, Chematica uses similar reactants, reducing the range of reagents, equipment and different synthetic paths needed to produce the library or selection of isotopically labelled compounds. Alistair Boyer, a synthetic chemist at the University of Glasgow, UK, notes that ‘any method of increasing efficiency in the laboratory is highly valuable, especially in the competitive field of drug development.’
‘This study showed something I didn’t know I lacked, but can very clearly see the future need of,’ comments Per-Ola Norrby who works on understanding and predicting selectivity in catalysis at AstraZeneca in Sweden. Norrby further highlights the utility of being able to broaden computational design to groups of similar compounds. Broadening the accessibility of complex retrosynthetic analysis to more researchers, and proving Chematica’s ability to achieve this, is something that Grzybowski is very keen on.
While his team work towards what he refers to as the ‘Holy Grail’ of synthetic tasks, Grzybowski is eager to invite top organic chemists to compete with Chematica in designing the best synthetic schemes to difficult products. ‘We are looking for challengers’, Grzybowski states, throwing down the gauntlet for a heavyweight competition of the best synthetic chemists and state-of-the-art retrosynthetic code.
References
This article is open access
K Molga, P Dittwald and B Grzybowski, Chem. Sci., 2019, DOI: 10.1039/c9sc02678a










No comments yet