Harnessing the power of the nitro group
Of all the chemical moieties found in natural products, nitro groups are among the least common. There are famous examples such as chloramphenicol (which also has the distinction of being the first antibiotic made by total synthesis, in 1949) but I wouldn’t need both hands to count them.
Despite the relevance of nitrogen in drugs, I’m also yet to work with a nitro compound in my career as a process chemist. Nitration of arenes is a great way to get nitrogen into a molecule, but the reagents are nasty, the exotherms are perilous and any nitro-containing intermediates have to be carefully tracked throughout a synthesis as they’re potentially mutagenic. In the academic realm, on the other hand, nitro groups are a pretty common sight in both the classroom and the lab. However, they’re most often thought of as an initial step in introducing an amine, and it’s rare indeed that you could say that strategic use of a nitro group was integral to a total synthesis. It’s also an underappreciated fact that nitroalkanes are as acidic as phenol, and due to their vulnerability to acids, bases, nucleophiles and reductants (to name just a few) it’s usually unwise to keep them around any longer than absolutely necessary.
Dawei Ma’s group at the Shanghai Institute of Organic Chemistry, China, have completed total syntheses of four members of the ever-popular gelsedine class of alkaloids using a clever Michael addition–aldol reaction combo to quickly assemble the dense tetracyclic core shared by this family. Key to pulling off this cascade is an alkyl nitro group that’s actually essential for both reactions and is somehow then carried through the entire natural product synthesis until its reduction in the penultimate step.
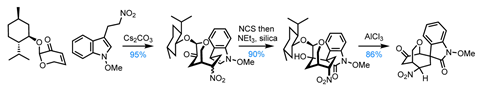
The route begins with the nitroalkane conjugate addition, which is rendered diastereoselective using menthol as a chiral auxiliary. Although chiral auxiliaries are often viewed as a bit dated these days, menthol is a supremely practical chiral building block and a kilogram costs less than a large round bottomed flask. Despite its apparent distance from the auxiliary, the new stereocentre on the cyclohexane ring is set with perfect stereocontrol, although for the time being the compound is a mixture of diastereomers at the nitro group.
Next, the indole is converted to an oxindole by oxidising with N-chlorosuccinimide, which kicks off a highly diastereoselective aldol reaction, effortlessly setting the challenging spiro quaternary centre. Interestingly, the stereochemical outcome of this reaction is controlled by the nitro group, which favours the desired transition state by hydrogen bonding to the enol form of the oxindole. With the cyclisation complete, a little base encourages the nitro group to equilibrate to its more stable equatorial form. A Lewis acid-mediated pinacol rearrangement then removes the chiral auxiliary, while simultaneously reconfiguring the bridged bicycle ready for completion of the natural product.
After a few oxidation state adjustments, all that remains is formation of the unusual azetidine ring system. The group bids a fond farewell to the nitro group, reducing it to an amine, which – with a little thermal encouragement – bites down onto the neighbouring epoxide, completing gelsmoxonine.
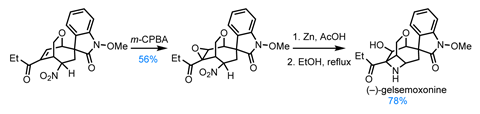
Through careful design and skilful use of the nitro functionality, the group has achieved not only the shortest synthesis of gelsemoxonine to date, but also four other challenging natural products – with nary a protecting group in sight.
References
1. P Wang, Y Gao and D Ma, J. Am. Chem. Soc., 2018, 140, 11608 (DOI: 10.1021/jacs.8b08127)











1 Reader's comment