Predicting protein structure doesn’t necessarily say much about function

Not so long ago, a list of ‘holy grails of chemistry’ like that recently compiled by Chemistry World might very probably have included ‘solving the protein folding problem’. It was widely believed that the ability to predict the structure of a protein from just its amino-acid sequence would be of immense value to the life sciences.
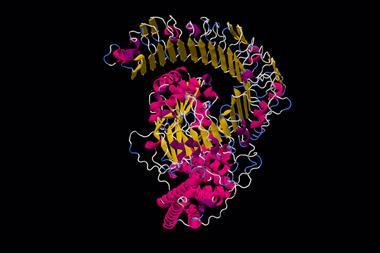
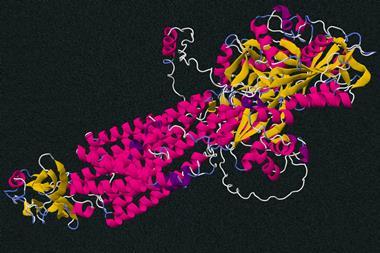
At the start of December, many media headlines announced what appeared to be the realisation of that goal. The artificial-intelligence company DeepMind has shown that their AlphaFold deep-learning algorithm can predict many protein structures from their sequence with an atomic-scale precision often comparable to that obtained from the best crystallographic analyses. It has been hailed as a major breakthrough. ‘It will change everything’, evolutionary biologist Andrei Lupas told Nature, while structural biologist Janet Thornton said the advance will ‘really help us to understand how human beings operate and function’. Some reports would have us believe cures for diseases such as Alzheimer’s (which stems from protein misfolding) are now just around the corner.
But such assertions have been contested. Some biochemists pointed out that the accuracy of prediction was not always so impressive and is in general unlikely to be accepted without experimental corroboration from, say, crystallography, NMR studies or cryo-electron microscopy. While the majority of predicted structures were within experimental resolution, one can’t tell a priori which are and which aren’t – so you need experiments to check. Also, it’s still not yet clear that the accuracy meets what’s needed for, say, finding drug candidates that might bind to the protein’s active site to block its function.
Unravelling the main issues
Others take issue with the notion that the method ‘solves the protein folding problem’ at all. Since the pioneering work of Christian Anfinsen in the 1950s, it has been known that unravelled (denatured) protein molecules may regain their ‘native’ conformation spontaneously, implying that the peptide sequence alone encodes the rules for correct folding. The challenge was to find those rules and predict the folding path.
AlphaFold has not done this. It says nothing about the mechanism of folding, but just predicts the structure using standard machine learning. It finds correlations between sequence and structure by being trained on the 170,000 or so known structures in the Protein Data Base: the algorithm doesn’t so much solve the protein-folding problem as evade it. How it ‘reasons’ from sequence to structure remains a black box.
If some see this as cheating, that doesn’t much matter for practical purposes. It will surely be valuable to deduce even a good guess at the structure from just the sequence. From that we can often make inferences about the protein’s function and the chemical mechanism of its mode of action. And ‘good enough’ predictions can be a useful starting point for refinement with crystallographic data.
But the idea that the protein-folding problem holds the key to understanding how gene sequences dictate cell function looks less compelling than it did a few decades ago. We know the real picture is much more complicated, for many reasons.
Tangled up
There’s more to enzyme action than correct folding. Many proteins are chemically modified after being translated on the ribosome: parts of the peptide chain may be crosslinked, and non-amino-acid groups such as porphyrins or metal ions are incorporated. Besides, knowing the structure doesn’t by itself tell you the function. Sometimes this can be deduced by analogy, or rather, homology: proteins with similar folds may have similar functions. But that’s not invariably true: proteins with very similar structures can behave in chemically very different ways, while very different folds can achieve similar transformations. There is no unique structure-function relationship.
What’s more, designing a ligand for a protein can be challenging even if you know its structure very accurately, partly because we don’t know all the rules of recognition – some depend, for example, on fine details of solvation at the active site. And for drug discovery the biggest hurdles are typically upstream from the identification of a potential molecular target – not least because it often proves to be the wrong target.
In any case, the picture in which protein function is determined by a unique and static crystal structure is known now to be far too simplistic. The dynamics might be crucial. Ligand binding typically involves some flexibility and adaptation at the active site – but more generally, the emerging view of protein function invokes the ensemble of conformations accessible to it: the statistical populations and occupancy times of the different dynamic states it can reach. What’s more, many proteins don’t have well-defined folded conformations at all, but contain ‘intrinsically disordered’, floppy parts of the peptide chain. That’s not nature being sloppy: the disorder and resulting flexibility seems to be functional. AI approaches may well identify which sequences are likely to be disordered, but that alone won’t help to understand their behaviour.
Finally, any deep-learning system is only competent within the bounds of its training set. We don’t know the size of the human proteome, but some estimates say that only around 5% of all human proteins have been crystallised and their structure determined. So the training data are likely to be biased towards the structures that are relatively easy to solve. Some researchers think there could be a systematic repertoire of protein structures that we just don’t know about.
None of this is to diminish the achievement of AlphaFold – and indeed we can anticipate that AI approaches might help tackle some of these caveats too. The real point is that we have long ago had to abandon the simple notion that the cell’s secrets are digitally encoded in any molecular sequence.
Not so long ago, a list of ‘holy grails of chemistry’ like that recently compiled by Chemistry World might have included ‘solving the protein folding problem’. It was widely believed that the ability to predict the structure of a protein from just its amino-acid sequence would be of immense value to the life sciences.
At the start of December, many media headlines announced what appeared to be the realisation of that goal. The artificial-intelligence company DeepMind has shown that their AlphaFold deep-learning algorithm can predict many protein structures from their sequence with an atomic-scale precision often comparable to that obtained from the best crystallographic analyses. ‘It will change everything’, evolutionary biologist Andrei Lupas told Nature, while structural biologist Janet Thornton said the advance will ‘really help us to understand how human beings operate and function’. Some reports would have us believe cures for diseases such as Alzheimer’s (which stems from protein misfolding) are now just around the corner.
But such assertions have been contested. Some biochemists pointed out that the accuracy of prediction was not always so impressive and is in general unlikely to be accepted without experimental corroboration from, say, crystallography, NMR studies or cryo-electron microscopy. Also, it’s still not yet clear that the accuracy meets what’s needed for, say, finding drug candidates that might bind to the protein’s active site to block its function.
Others take issue with the notion that the method ‘solves the protein folding problem’ at all. Since the pioneering work of Christian Anfinsen in the 1950s, it has been known that unravelled (denatured) protein molecules may regain their ‘native’ conformation spontaneously, implying that the peptide sequence alone encodes the rules for correct folding. The challenge was to find those rules and predict the folding path.
AlphaFold has not done this. It says nothing about the mechanism of folding; how it ‘reasons’ from sequence to structure remains a black box.
If some see this as cheating, that doesn’t much matter for practical purposes. It will surely be valuable to deduce even a good guess at the structure from just the sequence. From that we can often make inferences about the protein’s function and the chemical mechanism of its mode of action. And ‘good enough’ predictions can be a useful starting point for refinement with crystallographic data.
But there’s more to enzyme action than correct folding. Many proteins are chemically modified after being translated on the ribosome: parts of the peptide chain may be crosslinked, and non-amino-acid groups such as porphyrins or metal ions are incorporated. Besides, knowing the structure doesn’t by itself tell you the function: proteins with very similar structures can behave in chemically very different ways, while very different folds can achieve similar transformations. There is no unique structure-function relationship.
What’s more, designing a ligand for a protein can be challenging even if you know its structure very accurately, partly because we don’t know all the rules of recognition – some depend, for example, on fine details of solvation at the active site. And for drug discovery the biggest hurdles are typically upstream from the identification of a potential molecular target – not least because it often proves to be the wrong target.
In any case, the picture in which protein function is determined by a unique and static crystal structure is far too simplistic. The dynamics might be crucial. Ligand binding typically involves some flexibility and adaptation at the active site – but more generally, the emerging view of protein function invokes the ensemble of conformations accessible to it: the statistical populations and occupancy times of the different dynamic states it can reach. What’s more, many proteins don’t have well-defined folded conformations at all, but contain ‘intrinsically disordered’, floppy parts of the peptide chain. That’s not nature being sloppy: the disorder and resulting flexibility seems to be functional. AI approaches may well identify which sequences are likely to be disordered, but that alone won’t help to understand their behaviour.
Finally, any deep-learning system is only competent within the bounds of its training set. Some estimates say that only around 5% of all human proteins have been crystallised and their structure determined. So the training data are likely to be biased towards the structures that are relatively easy to solve. Some researchers think there could be a systematic repertoire of protein structures that we just don’t know about.
None of this is to diminish the achievement of AlphaFold – and indeed we can anticipate that AI approaches might help tackle some of these caveats too. The real point is that we have long ago had to abandon the simple notion that the cell’s secrets are digitally encoded in any molecular sequence.









No comments yet