Phenalenyl cations can act as both redox and Lewis acidic catalysts
A transition metal catalyst can be Lewis acidic to some extent, but it’s hard to imagine one with acidity comparable to classical inorganic Lewis acids such as AlCl3 or FeCl3. However, it’s easy to imagine the fun that could be had with a catalyst that is both redox active (capable of electron transfer) and highly Lewis acidic.
Given the apparent dearth of such molecules, organic chemists have focused on developing a multitude of dual catalytic reactions that use two distinct, yet chemically compatible, catalysts to mediate reactions that need activation on two fronts. As outstanding as these examples often are, when it comes down to it, they’re simply not that elegant – it’s a workaround.
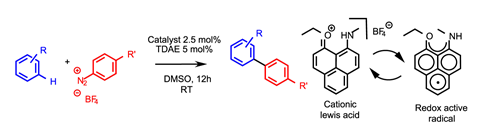
Fortunately, bringing style to the forefront, Swadhin Mandal and his team at the Institute of Science Education and Research in Kolkata, India, have developed a beautiful organic catalytic system that rolls Lewis acidity and redox activity into a single molecule.1 They have developed a phenalenyl cation (PC) that catalyses a formal cross-coupling reaction to generate biaryls from aryldiazonium salts and simple arenes that are functionalised at a carbon-hydrogen bond (figure 1). While the reaction itself is nothing to shout about, the underlying catalysis is a treat.
Perinaphthenone-derived phenalenyl cations are stable, delocalised systems, with 12 electrons distributed over 13 atoms, and have been known since the 1950s. Crucially, they have a relatively low-energy empty non-bonding molecular orbital. Adding one or two electrons to that orbital (to form a radical or anion) does not significantly compromise the stability of the molecule either, and hence comes with a low energetic cost.
The team has already shown that single electron reduction of the phenalenyl cation generates a redox active system capable of reducing aryl diazonium salts to generate nucleophilic phenyl radicals.2 They then proposed that benzene itself could be activated towards radical addition by forming a charge-transfer complex with the same phenalenyl cation that kick-started the reaction.
The team uses tetrakis(dimethylamino)ethylene (TDAE) to first reduce the phenalenyl cation, which in turn reduces the diazonium salt. While TDAE can, in theory, reduce the diazonium salt directly, UV-Vis spectrometry showed that electron transfer from TDAE to the phenalenyl cation is five times faster than to the diazonium salt, and is therefore it is likely to dominate. Then, using multiple analytical methods, the team confirmed the interaction and charge transfer between the phenyl coupling partner and the non-bonding orbital of the phenalenyl cation.
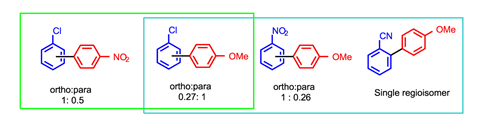
This simple and elegant system is effective at coupling a host of aryl diazoniums with benzene derivatives in dimethylsulfoxide at room temperature, although surprisingly there are many fascinating aspects of the reactivity that appear to have not been questioned: Why does it appear than the nucleophilicity of the aryl radical has an influence on regiochemistry of bond formation (figure 2; green box)? Or why do chlorobenzene and nitrobenzene generate regioisomeric mixtures when other similarly electron-poor systems such as carboxylate esters and cyano groups show excellent regioselectivity (figure 2; blue box)? And crucially – do these electron-poor arenes interact at all with the phenalenyl cation? It is not hard to imagine activation of such systems through their functional groups rather than the less pronounced π-electron cloud, or possibly no activation is required at all.
What is presented is a beautiful demonstration of the dual redox and Lewis acidic properties of aromatic molecules with vacant non-bonding molecular orbitals. There is plenty of potential to modify the catalyst structure to modulate its redox properties as a single electron transfer agent, or even create a stereo-defined environment to activate pro-chiral radical acceptors. A few too many questions are left unanswered and are due at least a passing comment, although at least this leaves Mandal and his researchers plenty of opportunity for digging into the details. I very much look forward to the next instalment.
References
1 J Ahmed et al, J. Am. Chem. Soc., 2018, 140, 8330 (DOI: 10.1021/jacs.8b04786)
2 J Ahmed et al, Chem. Sci., 2017, 8, 7798 (DOI: 10.1039/C7SC02661G)












No comments yet