In defence of name calling
All scientific fields have their jargon and technical terms, but organic chemistry is unusually complex. The organic chemist’s argot encompasses not only many strange and otherworldly nouns (themselves almost infinitely compoundable), but also geometric descriptors and locants, highly abstract concepts and endless shorthand for motifs and mechanism. Learning this language takes years if not decades, and a chemist’s fluency is usually a good indication of their experience and skill in the field.
Since my early days as chemist, I’ve been fascinated by one small part of this vernacular in particular: the named reaction. Like Richard Feynman, I painfully learned early on the difference between knowing the name of something and knowing something, but over time I realised how useful these terms of art can be. Not only do named reactions facilitate communication between chemists – imagine discussing a retrosynthesis where the mechanism of every proposed disconnection has to be described stepwise and verbatim – but they provide context and detail that help us retain abstract information.
Characters, patterns and connections are an invaluable aid to the human brain. Consider phrases such as ‘Catch 22’, ‘Hobson’s choice’ or ‘sword of Damocles’ – they act as linguistic shorthand, and add colour to our conversation and writing. Named reactions also satisfy humans’ desire for narrative – the late Terry Pratchett half-jokingly characterised us not as homo sapiens but as pan narrans – the storytelling ape. So I’m always keen to learn new named reactions and the stories behind them.
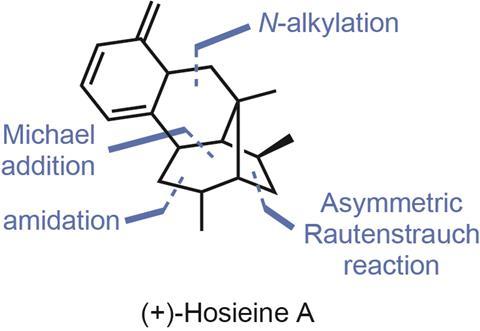
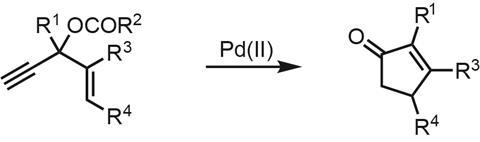
To my delight, a recently published total synthesis of the compact alkaloid hosieine A (figure 1) from John Wood and coworkers at Baylor University in Texas, US, introduced me to a powerful carbon–carbon bond forming reaction that I’d never heard of: the Rautenstrauch reaction.1 First reported in 1984, the original reaction isn’t much to look at (figure 2), using a palladium catalyst to rearrange enynes into simple cyclopentenones. However, later follow up studies showed that when enantioenriched starting materials are used, the chiral information can be transferred to the products with surprisingly fidelity.
A much more useful, but less-developed version of the reaction takes racemic starting materials and transforms them stereoconvergently into enantioenriched products through the use of a chiral catalyst, and it’s this version that the Wood group uses to prepare (+)-hosieine A. Taking a report by Dean Toste’s group using gold catalysts as initial proof-of-concept, the team sets about optimising the reaction. After scouring various ligand collections, they find that a chiral diphosphorus ligand (Biphep) gives the desired product in 90:10 er and good yield. Moreover, as expected, antipodal catalysts can be used to access either enantiomer of the product, and thence the final target. From here, the group forges the two remaining rings in short order. Michael-type addition of the ester to the newly-formed cyclopentenone under basic conditions closes the central piperidine, and impressively the single stereocentre in the starting material is able to direct the formation of three more in the product. The final ring is closed by reductive amination of the ketone with methylamine and sodium triacetoxyborohydride.
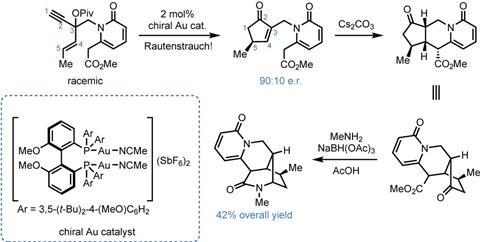
I’m impressed how well the Rautenstrauch reaction fits into this synthesis, taking a simple racemic starting material and giving rapid, enantioselective access to a tricky chiral cyclopentenone fragment that’s then able to direct formation of the remaining stereogenic centers. In fact, the entire sequence (figure 3) is run without any isolation of the intermediates until the final amide is formed. From here it’s a simple amide reduction with borane to get to the natural product, all in just seven steps. This is even more impressive when you realise that until this work, the shortest enantioselective route that had been published took over twenty steps to reach the target – and was by no means a bad synthesis.2
My thanks to the Wood group for so beautifully exemplifying a reaction that I’d never heard of!
References
1 J L Wood et al., Angew. Chem. Int. Ed., 2018, DOI: 10.1002/anie.201804076
2 R Hong et al., Angew. Chem. Int. Ed., 2015, 54, 10940 (DOI: 10.1002/anie.201505251)












No comments yet