
The LED zeppelin is flying high, but is it all hot air asks Karl Collins
Photoredox catalysis is a divisive topic. Not because the science is debatable, but because some feel it’s inescapable. They argue that the proliferation of photoredox publications is distorting the reality of progress in the field; incremental steps worthy of a mid-level journal are suddenly bumped up into the higher echelons of impact factors just because a blue light is involved. It’s true that some work certainly receives more attention than it deserves, and yes, there’s a lot of work with negligible value (or less), yet all that attention has its benefits too. Photoredox catalysis has become the glamorous cover model for synthetic chemistry, and we need one of those!
Photoredox is raising the bar for what can be considered a great synthesis
‘But science shouldn’t need to be sexy,’ protests the angry chorus. Thankfully, photoredox catalysis doesn’t just look great in pictures – the really good stuff is genuinely changing how we make molecules. In general it uses mild reaction conditions and relatively benign reagents, has good tolerance for functional groups, and is happy in a wide range of solvents, which means this chemistry is applicable to the kind of compounds that need to be made in the life sciences – polar molecules with basic centers. Naturally, not all of reactions work all of the time, but the best ones are applicable to a wider range of molecules that you can imagine. Trust me, I tried.
One of the most outstanding recent pieces of work comes from a newcomer to the field, Robert Phipps, from the University of Cambridge, UK. His team combined asymmetric counter ion catalysis and photoredox catalysis to solve the longstanding challenge of an asymmetric Minisci type reaction – the addition of a radical nucleophile to an electron deficient heterocycle. This is difficult because the radical’s prochiral centre inverts so quickly that it is hard to direct the reaction to produce a single enantiomer.
Phipps and his team started with the hypothesis that a chiral phosphoric acid would activate a basic heterocycle such as pyridine or quinoline by protonation of the nitrogen atom (Figure 1). As the chiral conjugate base should stay associated with the pyridinium cation, activation occurs in a chiral environment. Radical addition guided by hydrogen bonding interactions between a prochiral radical nucleophile – in this case derived from a redox-activated amino acid – and the chiral counter ion, should result in addition with enantiocontrol.

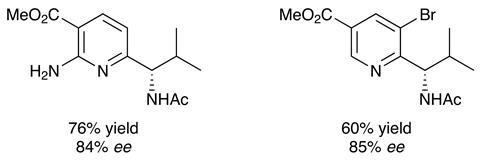
Starting with 4-methylquinoline, the researchers demonstrated that using 1mol% of a standard iridium based photocatalyst in combination with 5mol% of a chiral binol derived phosphoric acid they were able to synthesise the Minisci addition product in 93% yield and 96% ee. The racemic radical precursor was a redox activated N-acetyl phenyl alanine and the reaction proceeded at room temperature under irradiation with blue LEDs in 1,4-dioxane for 14h. The enantiomeric excess of the reaction was typically above 90% irrespective of the amino acid derivative – with alanine and tryptophan derivatives showing the extremes of the substrate scope. I’m sure it was a pleasant surprise to find that substitution of the quinoline was not required to ensure regioselectivity; quinoline itself yielded the 2-functionalised product with 89% yield and 96% ee.
Pyridines also work, although they need to be activated with an electron withdrawing substituent and the phosphoric acid catalyst has to be bulkier for high selectivities – but this is a moot point when the reward is access to such highly functionalised chiral pyridines (Figure 2). Isoquinolines are reactive, but the enantiocontrol is poor and imidazole derivatives failed to give isolable products.
This work is notable for several reasons: it merges two very contemporary fields of catalysis for the first time; it provides densely functionalised polar and basic building blocks – a dream for medicinal chemists; and it delivers enantioselective Minisci reactions. It’s an impressive display of photoredox catalysis and it deserves its moment in the spotlight.
Photoredox catalysis is no magic bullet – it has the practical and chemical limitations akin to all chemical reactions – but it is raising the bar for what can be considered a great reaction. Comparing photoredox catalysis to other revolutions in synthesis, such as cross coupling or metathesis, it is clear photoredox is special. Since 2010 it has progressed from being an academic curiosity to having industrial relevance; in just a few years and relatively few publications, it is already being integrated into the chemist’s standard array of synthetic tools. Maybe marketing is part of the story, but the chemistry works and consequently it is hard to argue against its cover star status.
References
R S J Procter, H J Davis and R J Phipps, Science, 2018, 360, 419 (DOI: 10.1126/science.aar6376)













No comments yet