Assessing a concept that has become messy

If you’re inclined to resist the popular assertion that chemistry is just applied quantum physics, you need do no more than invoke the notion of aromaticity. Of all chemistry’s messy, ill-defined concepts, none is more so than this. It shows that chemistry is in some ways closer to sociology or zoology: populated by individuals – molecules – that convenience impels us to classify and group without being quite sure if our categories are sound.
The very word betrays its shaky status, being one of the more obvious misnomers of the field. It stems, of course, from the central role of the benzene ring, the hexagonal core of a slew of organic compounds notable for their pungency by the time August Kekulé proposed the cyclic structure in 1865. Michael Faraday immediately noted the almond scent of benzene when he first isolated it in 1825. (He called it bicarburet of hydrogen, believing the ratio of carbon to hydrogen to be 2:1 because of the erroneous atomic weight then assigned to hydrogen.) Kekulé presented the structure in an odd ‘sausage’ format in his 1865 paper; only in 1872 did he show the familiar rings, noting that there were two equivalent configurations of the alternating single and double carbon–carbon bonds.
The very word betrays its shaky status, being one of the more obvious misnomers of the field. It stems, of course, from the central role of the benzene ring, the hexagonal core of a slew of organic compounds notable for their pungency by the time August Kekulé proposed the cyclic structure in 1865. Kekulé presented this structure in an odd ‘sausage’ format in his 1865 paper; only in 1872 did he show the familiar rings, noting that there were two equivalent configurations of the alternating single and double carbon–carbon bonds.
In the ring
It wasn’t until the quantum theory of chemical bonding in the 1930s that benzene’s electronic structure was clarified. Linus Pauling proposed that the two alternative Kekulé structures oscillate rapidly back and forth in a ‘resonance’ that accounts for the unusual stability of benzene: it is more stable than would be expected for any one of the Kekulé structures. Erich Hückel, meanwhile, offered another description based on his notion of molecular orbitals, in which the hexagon of σ bonds between carbons is supplemented by the continuous rings of π electrons from 2p orbitals overlapping above and below the plane of the atomic nuclei. These π orbitals may sustain circulating electrical ‘ring’ currents in an applied magnetic field, the effects of which account for the distinctive chemical shift of hydrogens attached to aromatic groups in NMR spectroscopy. That shift offers one way of assessing aromaticity.
The trouble is, it’s not unique in doing so. In fact, there are many such measures – based, for example, on bond lengths, electronic structure, energetics and chemical reactivity. And they aren’t all consistent, so the extent to which a molecular fragment is ‘aromatic’ depends on how you define it. Ostensibly tied to the quantum description of bonding, this putative feature of molecules is actually far more diffuse.
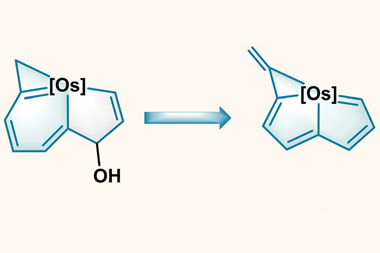
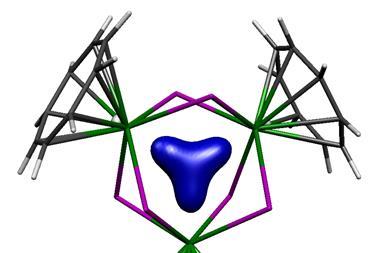
Arguments based on the symmetry properties of the electronic wavefunction that are needed to create a ‘closed circuit’ of mobile electrons led to the initial view that aromaticity was confined to planar ring systems with 4n+2 electrons, but that limitation has long since been abandoned. For example, ring-like molecules with conjugated bonds that have the single twist of a Möbius strip attain aromatic-like stability if they have 4n electrons. It’s now generally agreed that σ as well as π bonds can partake in aromaticity. And it can appear in three-dimensional systems too, especially involving main-group elements such as clusters of boron atoms but also including the cage-like fullerenes. The latter illustrate the complications of an aromaticity criterion based on energetic stabilisation – for how does one disentangle that from the strain energy of the curved framework?
It’s multiplying!
With so little agreement about what is and is not aromatic, the concept is open to extensions and modifications – and, some would say, abuses. Thus we see the appearance of ‘doubly aromatic’, ‘hyperaromatic’ and ‘superaromatic’ molecules. The latter, for example, was claimed for the macrocycle kekulene, made from 12 fused benzene rings in a hexagonal formation, until a recent single-molecule structural study using atomic force microscopy showed that the molecule does not after all contain two concentric, fully delocalised π rings but is more like six independently aromatic units welded together.1
Still the putative variety of types of aromaticity proliferates: spherical and cubic aromaticity, transition-state aromaticity, homoaromaticity, and more. It has got to the point where some chemists disdain the whole concept as irredeemably vague and sloppy. Others instead cry ‘Enough already!’ We should stop multiplying subclasses of aromaticity, says Miquel Solà of the University of Girona, and apply more self-discipline over the criteria used to identify it.2
Roald Hoffmann of Cornell University attributes aromaticity inflation to the ‘natural human tendency to want our molecular children to be exceptional’.3 Suppose we posit a molecule in which the occupied σ orbitals constitute a closed group – then let’s call it σ-aromatic! And yet, Hoffmann says, for some of these hypothetical molecules the supposed stabilisation applies to a structure that wouldn’t survive an instant in air at room temperature.
It’s not so much the hype that bothers him, but ‘the damage done to this beautiful, eminently chemical idea that I can trace back a century and a half.’ Messy or not, aromaticity surely stands for something – so we should be wary of debasing its currency.
References
1 I Pozo et al, J. Am. Chem. Soc., 2019, 141, 15488 (DOI: 10.1021/jacs.9b07926)
2 M Solà, Front. Chem., 2017, 5, 22 (DOI: 10.3389/fchem.2017.00022)
3 R Hoffmann, Am. Sci., 2015, 103, 18 (DOI: 10.1511/2015.112.18)








No comments yet