Prion diseases such as Creutzfeldt-Jakob are invariably fatal. But, as Dennis Rouvray reports, better insights into the protein chemistry involved are leading to new therapies
Prion diseases such as Creutzfeldt-Jakob are invariably fatal. But, as Dennis Rouvray reports, better insights into the protein chemistry involved are leading to new therapies
Many of the life-threatening new diseases that now confront us have arisen because of increasingly intimate contacts between humans and animals. Our progressive invasion of animal habitats and intensive farming methods have created conditions in which animal pathogens can jump the species barrier and become infectious to humans.
At present, roughly two-thirds of the 1400 infectious diseases we are susceptible to are known to originate from animal sources. But brand new diseases continue to be added to the list, some 30 having been identified over the past 50 years. Examples include AIDS and Ebola virus acquired from African primates, and SARS and avian flu from Asian poultry.
Britain’s infamous contribution was made in the 1980s when bovine spongiform encephalopathy (BSE), commonly known as mad cow disease, began to spread like wildfire through British cattle. By the 1990s it was clear that consumption of meat from infected animals was causing people to come down with variant Creutzfeldt-Jakob disease (vCJD), the human form of BSE. This and a dozen other similar diseases are designated transmissible spongiform encephalopathies (TSEs), because the brain of an infected organism takes on the appearance of a sponge.
Prions take the blame
From the outset, TSEs have shown some remarkable disparities from other infectious diseases. The induction period after infection is known to be exceptionally long; the victim enjoys normal health for many months or even years before being suddenly struck down with a devastating neurological disorder that invariably turns out to be fatal.
The infectious agent is also extraordinarily stable; neither irradiation with ultraviolet light nor treatment with formaldehyde nor boiling in caustic soda has any impact on its infectivity. Attempts to digest it with specific enzymes such as proteases or ribonucleases have also failed.
This seems to suggest that a virus is not involved, although a minority of researchers remain unconvinced and have continued to search for one. But their search is now viewed very much as a rearguard action, and the mainstream has moved on to embrace fully the once heretical notions of Stanley Prusiner of the University of California, San Francisco, US.
In 1982 he put forward the idea that the infectious agent consisted solely of protein, that it was a so-called proteinaceous infectious particle or prion for short. This idea, which as he later recalled ’set off a firestorm’, flew in the face of conventional biological wisdom that proteins cannot reproduce themselves without nucleic acids.
After a major campaign to counter this heresy failed to find any nucleic acids in prions, it became accepted that some hitherto unsuspected mechanism must exist for the spread of infectious diseases. When researchers eventually realised that the infected organisms were actually making the prions and that no biological principles were being violated, this field became ever more intriguing.
A small army of workers gravitated toward the investigation of prion diseases, even though vCJD - the main human form of prion disease - has an infection rate of about one in a million. Their combined endeavours over the past two decades have given us unparalleled new insights into the role of proteins as causative agents in human diseases.
We now know a great deal about the genes responsible for the production of prion proteins, the structure and nature of the prions formed, their folding and misfolding patterns, the part played by chaperone molecules in protein folding, and the reproduction of infectious prions in living organisms. This information is currently being exploited in a host of imaginative ways in the battle against the TSEs.
Prion proteins, PrP, which are naturally generated in the human body and normally reside on the membranes of cells, are encoded from a gene on chromosome 20. The primary and tertiary structures of the cellular form, PrPC, are well known (see box). Infectious prions, PrPSc, that are introduced into the body have essentially the same primary structure but a different tertiary structure.
When the two varieties come into contact, the infectious PrPSc prions begin to convert the normal PrPC prions into the infectious form. Although the process by which this takes place is not yet fully elucidated, it is thought that PrPSc prions exist as clusters of molecules that are effectively crystalline in nature. When PrPC prions encounter such a cluster they undergo a conformational change that enables them to fit and become integrated into it.
As Charles Weissmann of the MRC prion unit at the Institute of Neurology in London, UK, explains, ’depending on the structure of the crystal, the molecules that add to it will adapt to whatever the conformation is’. The PrPSc clusters break apart eventually and the infectious fragments migrate along peripheral nerves to the brain where they induce nerve cell destruction and the formation of microscopic vacuoles.
Disease attack
Armed with a knowledge of how prions operate, researchers are now formulating a multipronged attack on this group of diseases.
One approach that is unlikely to be viable is vaccination against TSEs. Because normal and infectious prions have the same primary structure, the human body is unable to recognise PrPSc prions as alien and so mounts no immune response against them.
This not only rules out vaccination but also conventional immunological methods to detect TSEs because infectious diseases are commonly diagnosed via surrogate markers, that is antibodies produced by the host organism when its cells are invaded. Other tests, such as that based on the polymerase chain reaction, depend on the identification of some kind of nucleic acid. None of these works for PrPSc prions, however, because the latter contain no nucleic acids and generate no antibodies.
These problems have been acknowledged in the US where a blue-ribbon task force has been set up under the chairmanship of Richard Johnson, professor of neurology, microbiology, and neuroscience at Johns Hopkins University School of Medicine in Baltimore, Maryland, US. In its 2004 report, Advancing prion science: guidance for the national prion research program, the task force admits that at present there is no truly reliable way of sterilising TSE-contaminated objects such as surgical instruments, that few of the basic materials employed in prion research have been standardised, and that no satisfactory blood tests are available for TSEs.
Creative thinking, however, is beginning to lead to ways around some of these major difficulties. For instance, it has proven possible to create antibodies that are active against prion diseases by injecting infectious prions into genetically engineered rodents incapable of synthesising PrPC prions. The infectious material stimulates the production of PrPSc antibodies, that can then be harvested.
Over two dozen companies are now manufacturing and patenting artificially made antibodies for prion diagnostics. The tests are performed on non-living tissues such as brain homogenates and the new antibodies preferentially tag PrPSc prions in a procedure described as conformation-dependent immunoassay. InPro Biotechnology, a company founded by Prusiner, is currently working with the prion business group at Beckman Coulter in Fullerton, California, US, to develop ever more sophisticated BSE diagnostic tests. Chris Neary, the group’s general manager, considers that an antemortem, blood-based diagnostic for TSEs could be the ’holy grail’ of prion disease research. Yet, given the considerable efforts on all continents to achieve this goal, he expects this to be accomplished in two to three years in the case of cattle and four to six years in the case of humans.
Work by Claudio Soto, now at the University of Texas medical branch in Galveston, US could hasten the realisation of this dream. His team has developed a so-called protein misfolding cyclic amplification procedure (PMCA) that uses sound waves to break up PrPSc clusters and multiplies many times over the opportunities for the PrPSc prions to convert PrPC prions into the infectious form. To date they have been able to increase prion concentrations by factors up to a millionfold, which is very near the level required for the development of a satisfactory blood test.
In a very promising recent study, the team used PMCA to screen blood from 18 prion-infected hamsters. The technique identified prions in 16 of these hamsters but found no prions in healthy animals.
Understanding folding
Extensive studies on protein folding carried out over the past decade have yielded a wealth of new insights into how this occurs under physiological conditions and within the rather cramped environment of a cell. It is now known, for instance, that the rate of protein folding is determined by the viscosity of the medium in which the protein is located and that the folding pattern is strongly influenced by ambient conditions such as pH and temperature.
In spite of the progress made so far, protein folding is not fully understood. Chris Dobson, head of a very active prion research group at Cambridge University, UK, considers that uncovering the mechanisms through which such processes take place is ’one of the grand challenges of modern science’.
Current thinking is that protein folding probably involves a stochastic probe by the polypeptide chain of the manifold different conformations it could adopt. It eventually arrives at a structure that is the most thermodynamically stable under the prevailing conditions and this is the structure that is usually found in the natural world.
Two control mechanisms operate to ensure that only properly folded proteins are formed in cells: molecules known as chaperones intervene and interact at various points to ensure the fidelity of the folding process, and any improperly folded proteins are promptly destroyed by proteolytic enzymes.
Future treatments
The manipulation of these control mechanisms will likely dominate the future course of therapeutic measures against the TSEs. Drug molecules are urgently needed that can meet at least one of three requirements: block the conversion of PrPC prions into PrPSc prions; facilitate the removal of any PrPSc prions formed; and diminish the toxicity of PrPSc prions.
Hitherto the focus has been very much on the first of these requirements, with efforts concentrated on either inhibiting the formation of PrPSc prions altogether or stopping them from aggregating once they form. PrPSc prions aggregate into rod-shaped fibrils when they reach the brain and remain infectious because the ends of the rods continue to convert PrPC prions into PrPSc prions.
Over the past three decades, numerous different compounds have been put forward that allegedly prevent this progression. These include polysulfated anions, dextrans, heparins, oligonucleotides, cyclic tetrapyrroles, anthrocyclines, porphyrins and diazo dyes. Tests on animals seem to indicate that most of these are effective provided they are taken at the time of infection but not if taken some time afterward.
More potent compounds that have also been tried include the bisacridines and especially quinacrine, which is known to bind to PrPC prions. But evidence for the efficacy of any of these drugs is largely anecdotal at present and the rigorous human clinical trials now being undertaken by the British Medical Research Council should soon provide us with some definitive answers.
Hopes of a breakthrough have recently been voiced by Mark Forman of the Center for Neurodegenerative Disease Research at the University of Pennsylvania in Philadelphia, US, who contends that ’there is a palpable sense of optimism among neurodegenerative disease investigators about near-term prospects for therapeutic advances in this field’.
Dennis Rouvray is a chemistry professor at the University of Georgia, Athens, Georgia, US
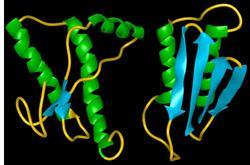
Human prion protein
The primary structure of the human prion protein, both in its benign and pathogenic forms, consists of a linear chain of 253 amino acids. This protein is synthesised in the endoplasmic reticulum of human cells, being encoded by the human gene PRNP located on chromosome 20. After translation, the prion migrates to the cell wall where it plays an important role in maintaining several cell functions.
Proteolytic enzymes such as protease K are able to digest the benign form in its entirety. However, the limited proteolysis that occurs as this form leaves the cell results in a circa 142 amino acid strand that is referred to as mature prion protein.
The folding of the linear prion protein can result in several different isoforms, the principal two being the normal or benign form and the abnormal or pathogenic form.
The normal form, PrPc where c denotes a cellular prion, consists of three α-helices at amino acids 144-154, 173-194 and 200-228, which are reinforced by a single disulfide bond connecting the amino acids 179 and 214. This prion is attached to the cell wall by a glycosylphosphatidyl inositol anchor and is glycosylated at the positions 181 and 197.
The pathogenic form, designated PrPSc, where sc stands for scrapie (the earliest known prion disease which occurs in sheep), is folded quite differently even though its primary structure is essentially identical.
The α-helical and β-sheet components are now reversed. The normal form has only two short b -strands that make up three per cent of the structure, while 42 per cent is α-helix. PrP 27-30 is 54 per cent β-sheet and 21 per cent α-helix. The transition from an α-helical-rich structure to a β-sheet-rich structure constitutes the fundamental difference in the two forms and is also responsible for their vastly different physiological interactions.
Additional information
- P Yam, The pathological protein: mad cow, chronic wasting, and other deadly prion diseases, 2003, Copernicus Books, New York, US
- R Erdtmann and L B Sivitz, Eds, Advancing prion science: guidance for the national prion research program, 2004, National Academies Press, Washington, DC, US
- S B Prusiner, Ed., Prion biology and disease, second edition, 2004, Cold Spring Harbor Laboratory Press, New York, US
- T Koster et al, J. Vet. Pharmacol. Therap., 2003, 26, 315
- A Smith, Ed., Insight: five protein misfolding articles, Nature, 2003, 426, 883
- J G Safar, Proc. Nat. Acad. Sci. US, 2005, 102, 3501






No comments yet