The history of anticancer drug temozolomide can be traced back over 30 years - and it all started with some novel nitrogen chemistry, says Clare Sansom
The history of anticancer drug temozolomide can be traced back over 30 years - and it all started with some novel nitrogen chemistry, says Clare Sansom
The pharmaceutical industry reserves the term ’blockbuster’ for its biggest-selling drugs - those that achieve sales of more than $1 billion during a calendar year. It is an accolade that very few drugs achieve. Of the many thousands of prescription drugs on the market, only about a hundred are blockbusters. They account, however, for about a third of all revenue earned from prescription drugs.

One of the latest drugs to gain blockbuster status is also the treatment of choice for some of the most malignant and intractable of brain tumours. Temozolomide, marketed in the US as Temodar and Europe as Temodal, made $1.02 billion (?650 million) for Schering-Plough in 2008. Temozolomide is a DNA alkylating agent, an unfashionable molecule in this era of targeted anticancer treatment. The story of its development is a remarkable mixture of scientific insight, serendipity, persistence and luck.
Temozolomide’s beginnings can be traced back over 30 years, to when medicinal chemist Malcolm Stevens set up a multi-disciplinary drug discovery laboratory in the pharmacy department at Aston University in central Birmingham, UK. In the 1970s, little was known about the nature and mechanisms of most proteins involved in cancer development. This was the era of ’chemistry led’ drug discovery. When a new research student, Robert Stone, joined the laboratory in 1978, on a PhD studentship part-funded by pharmaceutical company May & Baker, Stevens’ instructions to him were: ’Make some interesting molecules’.
Interesting molecules, to Stevens, were reactive heterocyclic ring systems rich in nitrogen atoms. Two strands of nitrogen chemistry led to the eventual discovery of temozolomide’s nucleus, the imidazotetrazine ring system. Firstly, by the late 1970s, it was recognised that some triazenes - molecules containing a linear chain of three nitrogen atoms - had anti-tumour activity. One triazene, dacarbazine (DTIC), was on the market against malignant melanoma, (and is still in clinical use); it is a pro-drug, forming the alkylating agent MTIC (5-[3-monomethyl-1-triazeno]imidazole-4-carboxamide) in vivo.
The second set of nitrogen-rich molecules inspiring Stevens were the tetrazine heterocycles. By the time Stone arrived at Aston, the group was exploring bicyclic ring systems with ’bridgehead’ nitrogen atoms between the rings. Stevens read a paper by G?nter Ege and Karlheinz Gilbert at the University of Heidelberg, Germany, describing a new synthetic route to fused tetrazine ring systems via the interaction of diazoazoles and isocyanates, and decided to try the method with his own molecules. This synthesis produced the first imidazotetrazinones - which, like the triazene drugs, contains three consecutive nitrogen atoms, although this time in cyclic form. ’We formed a brand new ring system that had never been seen before, and made a series of variants with different substituents’, remembers Stevens. ’We had a lot of chemistry to do, to determine their structure and physical properties, and, particularly, if they had any interesting pharmaceutical activity.’
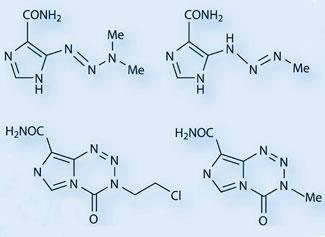
The first structure Stone synthesised contained a chloroethyl group, and was also found to have extremely interesting anti-tumour properties. Another PhD student in Stevens’ group, Neil Gibson - along with Aston academics John Hickman and Andreas Gescher - showed that it was effective with a single dose against almost all contemporary mouse models of cancer. Not surprisingly, the Aston group thought they had found the elusive ’magic bullet’ against cancer. The compound was given a lab name, azolastone, and preparations were made for a Phase I clinical trial through the Cancer Research Campaign (CRC; now Cancer Research UK). ’It was a great name, incorporating the azo group, the name of the lab (Aston) and of the student who synthesised it’, says Stevens.
Reality check
The bad news started almost immediately, with a controversy over the compound’s name. Carl Schwalbe, a crystallographer colleague, remembers what happened next. ’May & Baker thought the name too similar to the common antihistamine, azelastine, and made us change it. It was understandable - the idea of a patient being mistakenly prescribed a toxic anticancer drug for hayfever gave us nightmares - but we were sorry to lose azolastone. And I remember Malcolm being furious that the alternative name, mitozolomide, seemed to have no chemical rationale behind it,’ he says.
More serious disappointment was to come. The published Phase I clinical trial of mitozolomide revealed disappointing results. The molecule that had proved so successful in mice was found to be extremely toxic in humans, producing profound bone marrow toxicity (thrombocytopenia). An unkind competitor even named the molecule ’Azo-last-one’.
Those poor results could have been the end of the imidazotetrazinone series. May & Baker eventually dropped out of the programme, wanting nothing more to do with such cytotoxic drugs, so Stevens appointed Cancer Research Technology (CRT). Even at Aston there was no consensus as to how to proceed. Another colleague, John Slack, remembers meetings to decide which candidate drugs to put forward for clinical trials. ’According to our colleague Simon Langdon, there was only one other imidazotetrazine that demonstrated anti-tumour activity, and that was the methyl derivative: temozolomide. There wasn’t much support for it, as it was likely to be "just another toxic DNA alkylating agent" - Malcolm [Stevens] was the only one of us who was prepared to champion it into clinical trials.’

Mitozolomide’s severe bone marrow toxicity had been associated with its ability to cross-link DNA. The replacement of its chloroethyl group with the methyl group of temozolomide produced a compound that was less active but gave no possibility of cross-linking DNA; it was, therefore, expected at least to be free of that toxicity. It might, the group proposed, ring-open in vivo to form an alkylating agent like DTIC, but under chemical rather than metabolic control. It was on that basis that temozolomide was submitted to the CRC’s newly launched Phase I / II committee for consideration as a clinical candidate.
The entire project might have been stalled before then, however, by events thousands of miles away. Stone’s original synthesis involved methyl isocyanate, the compound that caused the Bhopal disaster in India in December 1984. For some time after that it was almost impossible to source this ’pariah molecule’, and the project could only go forward at all with bulk supplies of both methyl isocyanate and temozolomide. Fortunately, May & Baker had manufactured enough temozolomide for pre-clinical and Phase I studies, and the formulation and development work was undertaken at Aston by an academic group funded by the CRC. The Stone synthesis is still used to manufacture temozolomide today.
Dose dependent
The first Phase I trial, testing single doses of temozolomide in patients with advanced cancer in different sites, yielded no positive responses. One of the clinical investigators, Ed Newlands of Charing Cross Hospital, London, then suggested a change of dose schedule. Giving temozolomide on five consecutive days changed the picture completely. An unusual number of positive responses for a Phase I trial were recorded, mainly in patients with brain tumours. ’One or two Phase I patients saw almost biblical cures [with multiple doses of temozolomide], although this is not indicative of reliable clinical efficacy,’ says Sally Burtles, director of drug development at CRC and now Cancer Research UK. ’These were glioma patients who had been bed-bound, but became able to get up, go about their normal business, and even go on holiday.’ After the initial study, CRC took the compound into Phase II studies in glioma and melanoma to demonstrate its activity more clearly.
But if this very promising molecule was to progress beyond early Phase II, Cancer Research Technology needed a partner from the big pharma sector. ’We set off on a "roadshow" round the US, presenting the early clinical data to potential partners. The drug was eventually licensed to Schering-Plough, who still markets it,’ says Stevens.
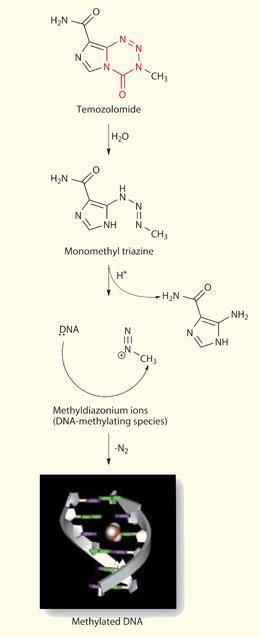
While temozolomide was progressing through the phases of clinical trials to registration, academics were continuing to elucidate the details of its mechanism of action. Phil Lowe, a PhD student working with Schwalbe, had already determined the crystal structures of both mitozolomide and temozolomide, confirming the molecular configuration that Stevens had proposed. ’Computational chemistry, along with an analysis of hydrogen bonding patterns, yielded some unexpected information,’ says Schwalbe. ’We found that the carbonyl carbon atom in the ring had an unusually high positive charge, supporting a mechanism in which the ring system is broken down at this position.’ Experiments by biochemist Mike Tisdale established that temozolomide is a pro-drug, a ’molecular delivery device’ that is stable under acid conditions but ring-opens at physiological pH to methylate, specifically, oxygen O6 of guanine residues, particularly those within runs of guanines.
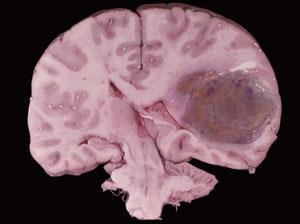
Temozolomide was first licensed in 1999, initially as a second-line treatment for glioblastoma multiforme, the most malignant grade of glioma. The indications for which it can be prescribed have been increasing since, along with its sales. It is now given with radiotherapy as the first line treatment for glioblastoma multiforme, and a second line treatment for other malignant gliomas after relapse. Jeremy Rees, consultant neurologist at the UCL Institute of Neurology, National Hospital for Neurology and Neurosurgery, London, explains how it has altered the outlook for these very sick patients. ’When given with radiotherapy, temozolomide has improved the two-year survival rates for these glioma patients from 10 per cent to 25 per cent and the four- to five-year survival rate has improved from almost negligible to 10 per cent.’ Rees is currently involved in a Phase III Cancer Research UK trial comparing temozolomide against radiotherapy as a first-line treatment for lower-grade, less malignant gliomas.
Although temozolomide was developed before the target-driven approach to drug design became fashionable, it does have a very distinct molecular target - the O6 oxygen atom of guanine residues within DNA, particularly those within poly-guanine sequences. It can therefore be expected to interact with proteins that repair damaged DNA. This can be used both to predict the response of individual patients to temozolomide, and to develop drugs that might act synergistically with it.
Doubling up
The enzyme 6-O-methylguanine-DNA methyltransferase (MGMT) is a DNA repair protein that removes methyl groups from the O6 position of guanine and can thus ’undo’ the action of temozolomide. Like many proteins, MGMT can be ’switched off’ or silenced by methylation of DNA residues in its promoter region, upstream of the coding sequence.
Recent clinical trials have suggested that those glioma patients with tumours in which the MGMT gene has been silenced by promoter methylation are more sensitive to temozolomide given with radiotherapy. One study showed clinical response to temozolomide in 68 per cent of glioma patients with MGMT silencing compared to 25 per cent of patients with active MGMT. It is possible that giving temozolomide with an MGMT inhibitor could improve the response of these patients, if one could be developed. However, one of the advantages of temozolomide that contributes to its premier position in the market is that it can be delivered orally. Many drugs for advanced cancer cannot be formulated for oral delivery and must be given in hospital: co-prescribing temozolomide with any non-oral drug would be less convenient for the patient and so would detract from the drug’s significant advantage.
Temozolomide has proved a very significant advance for many brain tumour patients, but it is not universally successful, and it can only lengthen life, not cure the cancer. Rees still describes brain tumours as ’a core area of unmet medical need. we still need new treatments.’ So, where will ’the next temozolomide’ - or even the next blockbuster anticancer agent - come from? It may be from the same stable. Stevens, now an emeritus professor at the University of Nottingham, is still developing temozolomide analogues and molecules with similar, nitrogen-rich, heterocyclic structures that might have even better antitumour activity. He is now also chief scientific officer of an innovative drug discovery company, Pharminox, which has further molecules with novel mechanisms in late stage pre-clinical development and Phase I clinical trials.
Above all, the temozolomide story shows the importance of collaboration in drug development. It was a textbook example of a development strategy, led by chemistry, that also involved disciplines as diverse as crystallography, pharmacology and pharmaceutics, and, once the importance of the DNA methylating agents was discovered, molecular biology and pharmacogenetics. And it shows the importance of leadership. ’Every drug discovery group needs someone who believes in their molecules and will champion them through difficult times. Malcolm Stevens was just such a champion for temozolomide,’ says Burtles.
Clare Sansom is a freelance science writer based in London and Cambridge, UK.
Further Reading
M F G Stevens, Temozolomide: From cytotoxic to molecularly targeted agent. In: S Neidle, Cancer Drug Design and Discovery, p157-172. New York: Academic Press, 2008






No comments yet