A reaction confined to the lab for 25 years could find favour with industry after chemists retune the catalyst
An asymmetric catalysis reaction limited to laboratory syntheses has received a makeover that could see it used on a large scale by drug-makers.
Daniel Lim and Don Coltart, from Duke University, North Carolina, have adapted chiral catalysts to tackle drawbacks of the reaction creating chiral ketones with alkyl groups on their neighbouring (alpha) carbon atoms. Such structures are found in many natural products and pharmaceuticals.
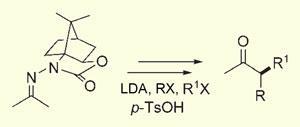
’Ketone alpha-alkylation is fundamental to organic synthesis, but only one effective asymmetric version of this transformation applicable to acyclic systems is available,’ the researchers explain. The traditional method, introduced over 25 years ago by Dieter Enders, uses chiral hydrazine auxiliaries that latch on to ketones, forming hydrazones. The auxiliaries ensure that only one enantiomer is formed when a hydrazone, deprotonated at its alpha carbon, attacks alkyl groups.
But this reaction, though it’s been used in many total syntheses, has a number of drawbacks. Deprotonation is very slow; the alkylation requires very low temperatures (-110?C to -78?C); and recycling the auxiliaries uses harsh conditions - such as reaction with ozone or the carcinogen methyl iodide - that can affect other parts of a molecule.
Coltart and Lim found that substituting conjugated electron-withdrawing groups onto the chiral hydrazine auxiliaries (forming N-amino cyclic carbamates) appears to make the hydrazones more stable, and allows the reaction to take place faster and in milder conditions. The alkylation took place in excellent yield and stereoselectivity at temperatures up to 0?C, and the auxiliaries could be fully recycled with a brief acid treatment. The hydrazone could even be alkylated twice on the same alpha carbon, because the auxiliary appears to override the selectivity of the deprotonating agent, lithium diisopropylamide (LDA).
’This is an impressive piece of work,’ says Derek Tan, a synthetic organic chemist working at Memorial Sloan-Kettering Cancer Centre, New York. ’The reactions are efficient, highly practical, and scalable and should be very useful for both academic and industrial chemists.’
Coltart hopes the new method will see the reaction become available to the pharmaceutical industry. He points out that drugs such as Zofran (marketed by GSK) and Aricept (Esai/Pfizer) are chiral ketones of the sort his method produces, but are marketed in racemic form - probably because they can’t be economically synthesised as single enantiomers. Larger drug molecules with chiral ketones that are marketed as single enantiomers, such as erythromycin, are prepared by fermentation.
’Importantly from a pharma point of view, the methodology is fast and simple and seems to be amenable to large scale synthesis,’ comments Christoph Gaul, who works for Swiss-based pharmaceutical company Novartis.
Coltart says the team has only tried the reaction on a 7gram scale so far. ’We’re very confident this will work at larger scales - it’s remarkably easy chemistry,’ he says. The researchers have applied for a patent on the process and Duke are exploring licensing the technology to pharmaceutical companies.
Richard Van Noorden
Enjoy this story? Spread the word using the ’tools’ menu on the left.
References
Angew. Chem. Int. Ed., 2008, DOI:10.1002/anie.200800848






No comments yet