High-throughput calculations pluck out selective alloys to replace palladium
In an impressive demonstration of the power of theoretical methods, researchers have used computer calculations to discover cheap alloys that selectively catalyse an industrial reaction better than the current expensive systems.[1]
Researchers led by Jens N?rskov at the Technical University of Denmark in Kongens Lyngby used density functional theory (DFT, which predicts electron density based on the principles of quantum mechanics) to find better ways of removing ethyne (HCCH) from a stream of ethene (H2C=CH2) used to make polymers.
The ethene produced by steam crackers usually contains about 1 per cent ethyne, a contaminant which must be removed by hydrogenation. But the process has to be selective, in order not to wastefully hydrogenate ethene to ethane. N?rskov explains that an expensive palladium-silver system is currently used as an industrial catalyst for the process, with the silver added to make sure that ethene desorbs from the surface faster than it can be hydrogenated.
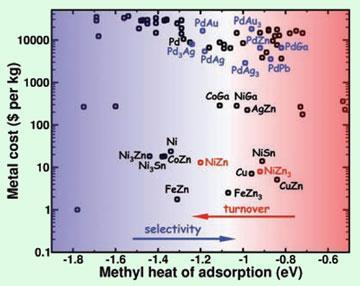
N?rskov’s team used DFT to quickly screen about 70 alternative bimetallic compounds, comparing their cost, stability, activity and selectivity of ethyne hydrogenation. They picked out a nickel-zinc alloy that performed well, and after synthesising and testing it with an oxide support, they showed it was indeed highly stable and selective for the required reaction.
’This shows not only that our theoretical descriptions of these reactions are correct,’ says N?rskov, ’but also that we can use DFT to predict something we don’t know in order to find new materials.’
Comprehensive catalysts
Density functional theory calculations have been used for about 15 years to calculate the structure and properties of metals and metal oxides in order to explain how molecules are reacting on them - but it’s only in the past few years that researchers have tried to use such computational methods to design better catalysts, says Jeff Greeley, who works at the Center for Nanoscale Materials, Argonne National Laboratory, Illinois, US.
N?rskov’s group has led the way with the first few examples of successful theoretical catalyst design.[2] But the latest study shows the most comprehensive approach yet, says Greeley, because it incorporates a study of catalyst selectivity.
The key to the team’s success was cutting down on computation time by finding that simply determining how effectively a methyl group clung to the surface of the alloys was a good approximation to the behaviour of more complex molecules, explains Matt Neurock, of the University of Virginia, Charlottesville, US. ’This is a very elegant study,’ he says.
’From knowing there are good systems out there to developing a real catalyst is an enormous step, but we are of course taking this further,’ says N?rskov. Ten months ago he and colleagues launched a company, Computational Materials Design ApS, which takes on research contracts from industrial partners to improve catalysts, using the computational high-throughput methods the group has developed.
Richard Van Noorden
Enjoy this story? Spread the word using the ’tools’ menu on the left.
References
2 J Greeley et al, Nature Materials, 2006, 5, 909






No comments yet