Richard Corfield explains how stable isotopes are helping to pinpoint geologic time.
Scientists have known since at least the 1950s that certain species of stable isotopes show systematic variations in abundance through time. However, it is only in the past decade that this finding has consolidated into a new branch of geology known as ’stable isotope stratigraphy’. This itself is part of an emerging larger discipline that uses elemental and molecular variations to identify different intervals of the geological record and which is known simply as ’chemostratigraphy’.
The initial focus on stable isotopes was largely a historical accident generated by the interest in unstable isotopes during World War II and at the start of the Cold War. First to discover these variations was the Nobel laureate and Manhattan Project alumnus Harold Urey. Urey was interested in finding a way of measuring temperatures in the geological past and he reasoned, on the basis of physical kinetics, that proportionally more of the light isotope of oxygen, 16 O, would be incorporated in shell material with a rise in temperature. This would be due purely to the fact that 16 O has greater vibrational energy than the heavier isotope 18 O, and so is more easily incorporated into a calcite lattice such as the shell of a mollusc.
Urey’s colleague Cesare Emiliani took the idea further and applied it to tiny, single-celled foraminifera that live in the surface waters of the ocean and which accumulate in vast numbers on the sea bed. Over eons these form vast drifts of sediment that can be sampled by deep sea coring.
The huge advantage of cores (compared with land or ’outcrop’ sections) is that the sediments are well ordered in time. It is therefore possible to be fairly sure that the sequence of events is accurately recorded when going from older to younger sediments. Emiliani was able to start to take Urey’s idea to its ultimate conclusion, the mapping of temperature change (ie climate) through geological time.
But it soon became clear that Urey’s idea was an oversimplification - oxygen isotope values do reflect temperature, but there is a contaminating influence: the growth and decay of the great ice sheets through glacial-interglacial cycles. When ice sheets grow they incorporate water that has been evaporated in the low latitudes and carried to the poles in the form of water vapour which is then precipitated. Evaporation favours the light isotope of oxygen and so polar ice has proportionally more 16 O than the seawater left behind. Hence foraminifera growing in the oceans during glacial intervals will already have a heavier 18 O signature than during interglacials. Conversely, during interglacials, both warmer temperatures and less ice equals more 16 O in fossil forams.
Investigations of foraminifera living on the sea bed (benthics), where temperatures are supposedly constant, revealed that they too showed large variations between glacial-interglacial intervals. This in turn suggested that changes in ice volume were far more important than temperature. However, for the purposes of chemostratigraphy this debate is academic; what is important is that both planktonic and benthic fossil foraminifera show systematic variations in time that reflect the growth and decay of ice-caps. A way had been found of unambiguously identifying times of glaciation in the fossil record.
Today, oxygen isotope stratigraphy has burgeoned into one of the most widely used analytical techniques in the palaeontological arsenal. To date researchers have identified more than 100 oxygen isotope ’stages’ and there seems no reason why this technique should not be extended to the beginning of the Cenozoic (Fig 1).
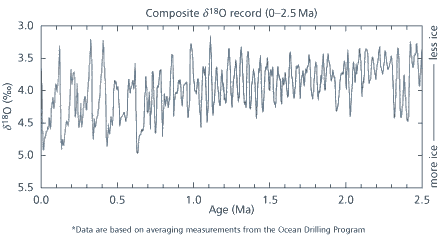
The discovery that these oxygen isotope variations occur with a regular cyclicity was one of the most important breakthroughs of the 1970s. In a seminal paper published in Science in 1976, Jim Hays, John Imbrie and Nicholas Shackleton proved beyond doubt that the cycles of glacial-interglacial ages were controlled by variations in the Earth’s orbit around the sun. The theory had originally been proposed by an obscure Yugoslavian astronomer named Milutin Milankovitch in the early years of the 20th century.
When the Earth’s orbit is in such a configuration as to minimise the amount of sunlight hitting the land masses of the northern hemisphere, greater amounts of ice and snow persist in the high latitudes during the summer months. Together with the increased reflection from the snow-bound Earth, eventually this plunges the world into an ice age. The reciprocal of this orbital configuration promotes interglacial ages.
The importance of this link between a metronome in the heavens and oxygen isotope variations in deep sea sediments cannot be overestimated for it has allowed, by turning the idea on its head, the ultra-accurate calibration of time. Each glacial-interglacial cycle has a invariant duration that must be reflected in the length of the oxygen isotope cycles in deep sea cores. Knowing this relationship has allowed geologists to recalibrate the timing of the crucial magnetic polarity reversals in the cores that, in turn, are the pegs on which hang the dating of fossil extinction or origination events. So now, when a palaeontologist identifies one of these datums in a deep sea - or indeed land section - the age of that rock layer is known with a previously undreamed of accuracy. This new discipline - astrochronology - is probably the supreme achievement of chemostratigraphy.
Bottom drawer data
Another type of isotope data began to be collected very soon after the oxygen isotope revolution of the 1950s started. Since CO2 contains both oxygen and carbon atoms it was a fairly trivial matter to acquire 13 C data from the same sample. The three most common species of CO2 are 12 C 16 O 16 O, 13 C 16 O 16 O, and 12 C 16 O 18 O. These have atomic masses of 44, 45 and 46 respectively and are easily separated in the flight tube of a mass spectrometer.
In the early days of oxygen isotope thermometry the most common MS instrument was the so-called ’dual-collector’ machine which had two Faraday cups for collecting the ionised CO2 molecules as they came screaming down the flight tube from the source. It was a simple matter to retune the accelerating voltage of the machine so that the 13 C beam was captured by the second collector. In this way carbon isotope data were collected along with the oxygen isotope data although at that time there was no clear appreciation of their usefulness - at least in a palaeoceanographic context. All that was known was that more of the light 12 C isotope was incorporated into the organic material formed by photosynthesis and that this left the surrounding seawater progressively more enriched in the heavy isotope of carbon, 13 C.
It was this relationship with photosynthesis that precipitated another revolution in chemostratigraphy in the 1970s, on the back of emerging concerns about the possible role of greenhouse gases like CO2 in controlling climate change. The intensity of photosynthesis occurring in the surface waters of the oceans controls the partial pressure of CO2 in the atmosphere above. Scientists soon realised therefore that the 12 C to 13 C ratio indicated variations in the partial pressure of atmospheric CO2 in the geological past.
The more green plants and photosynthesising bacteria there are, the more 12 C is locked up in organic material and transported away from the sea surface, and the more 13 C is incorporated into the shells of planktonic foraminifera precipitating CaCO3 from seawater. Deep water benthic foraminifera do not register these fluctuations in productivity, so that the difference between these forams calibrated against the recent past gives a proxy for the partial pressure of CO2 over geological time. This ’difference’ approach removes the effect of changes to the gross δ 13 C composition of the ocean which takes place on longer timescales.
Proof of the accuracy of the technique was its near perfect match with the record of CO2 change during the Pleistocene retrieved from ice cores at Vostok station in Antarctica (Fig 2). The crucial advantage of the carbon isotope difference technique was that it was not limited to the depth that an ice core could penetrate but, like the oxygen isotope technique, only by the recoverable depth of deep sea cores.
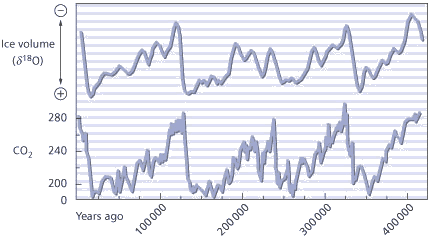
When it was found that the Pleistocene δ13 C changes occurred with a cyclicity as regular as those of the oxygen isotope record the search was on for a link between the two. Was there any relationship between the changes in atmospheric CO2 concentration implied by the regular cycles in δ13 C and the changes in ice volume implied by the variations in δ18 O?
The breakthrough came in the early 1980s when Nicholas Shackleton of Cambridge University showed, using spectral analysis, that the variations in CO2 concentration led ice-volume change but lagged the Milankovitch variations. The conclusion was inescapable: CO2 change was the mechanism by which changes in the Earth’s orbit triggered and terminated ice ages. Carbon dioxide was nothing less than the amplifier that turned subtle changes in the Earth’s orbital geometry into ice ages or warm periods. Any lingering doubts about the relationship between greenhouse gases and climate change were blown away in one fell swoop.
Time dependence
Since those heady days of the 1980s the relationship between oceanic productivity change and carbon isotope variations has been explored in more detail and is known to be heavily timescale dependent.
At one of end of the scale, δ13 C variations can be linked to high-frequency mechanisms such as changes in the Earth’s orbit around the sun, while at the other end (> a million years) long-term changes in productivity and the partitioning of organic carbon between the atmospheric, oceanic and terrestrial carbon reservoirs exert a profound effect. Nowhere is this better seen than in the period of Earth history known as the Palaeocene, a 10 million year interval that started immediately after the mass extinction that killed off the dinosaurs (the so-called K/T boundary) and which ended with the oceanic methane ’burp’ known as the Palaeocene-Eocene Thermal Maximum or PETM.
In general carbon isotopes are more useful than oxygen isotopes for stratigraphic purposes in the remote past (broadly, >10 million years before present). This is because they are more resistant to diagenesis: the tendency of the original isotopic ratio to be altered by post-depositional processes such as through-flow of meteoric water. Despite this, there are still limitations to the use of carbon isotopes as stratigraphic tools, partly because they too ultimately succumb to diagenesis in rocks that have been deeply buried or cooked and partly because the residence time of carbon in the ocean is not significantly longer than the mixing time of the ocean.
This means that the possibility always exists that different parts of the world’s oceans - and therefore the sediments and ultimately rocks derived from them - may have different carbon isotope ratios. By the mid-1980s therefore interest was focusing on yet another isotopic system - strontium - that does not suffer from this disadvantage because its residence time in the ocean is very long indeed compared to the mixing time of the oceans and also because the isotopic ratios are solely controlled by very long term processes.
For all the utility of chemostratigraphy, the best results are obtained when the various proxies are combined with other types of stratigraphic information. The most important of these is biostratigraphy - the succession of fossils in the rock record. Biostratigraphy is qualitatively different to chemostratigraphy because it is an ordinal process that never repeats itself. The evolution and extinction of life forms is, after all, unique; the dinosaurs, for example, will never evolve again.
Chemostratigraphy, in contrast, is iterative - there is no unique signature to an oxygen, carbon or strontium isotope ratio. Yet chemostratigraphy, the signatures of time, have transformed the way that we recognise the passage of eternity in the geological record.
Source: Chemistry in Britain
Acknowledgements
Richard Corfield is an isotope geochemist and science writer in the department of earth sciences, Oxford University, where he directs the stable isotope laboratory.
1. Perfect Palaeocene timing
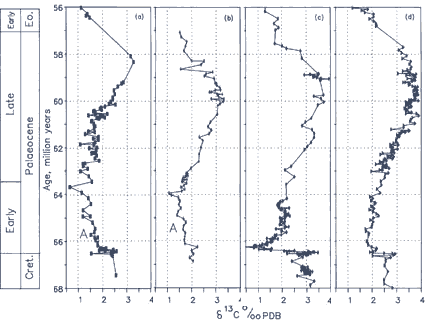
During the Palaeocene carbon isotope values rise steeply to a maximum at about 60 Ma (million years before present) then decline toward a minimum in the early Eocene (see Fig below). It seems that the meteorite collision that killed off the dinosaurs also destabilised the prevailing controls on carbon isotope change by killing off most of the oceanic plankton that control the partitioning of carbon isotopes between the atmospheric and marine reservoirs. As plankton evolution began again in the immediate aftermath of the catastrophe, CO2 was pulled out of the atmosphere and sequestered - probably in the form of organic carbon - in the deep ocean.
Unlike the Pleistocene this process was unregulated by Milankovitch cyclicity and continued for more than four million years. The peak in δ13C values at about 60Ma represents the time of maximum atmospheric CO2 depletion and it is no surprise therefore that at this time the seas near Antarctica began to cool, accelerating the formation of cold, deep waters there. What stopped this trend is unknown but after about 60 Ma δ13C values began their decline, signifying an increase in atmospheric CO2.
From the stratigraphic perspective the Palaeocene carbon isotope maximum is an enormously useful marker that identifies rocks of Palaeocene age even in the absence of other dating clues. The figure (below) also shows a comparison of the δ13C record from four sites: two from a rock section outcropping on land in central Italy, one from a deep sea drill hole the southeast Atlantic and the other from a drill hole in the central Pacific. The striking thing about all four curves is their similarity which shows that the Palaeocene carbon isotope event was a truly global phenomenon.
A single example serves to illustrate the importance of this point. In early 1990 Leg 130 of the international Ocean Drilling Program retrieved carbonate sediments from very deep below the ocean floor of the Ontong Java Plateau in the western equatorial Pacific. This rock had been so compacted and heated that all the microfossils - which would ordinarily have served to identify age - had been cooked and compressed into unidentifiable husks within a rock fabric so hard that it rang like a bell when tapped with a hammer. Yet a dark layer lower in the core had been tentatively identified as the K/T boundary. In my group’s laboratory at Oxford we analysed the barren sediments immediately above this layer and immediately retrieved the characteristic carbon isotope signature of the Palaeocene period. In the absence of other clues we still knew exactly where we were in time.
2. Strontium dating
Strontium-87 is a radiogenic isotope formed by the α-decay of 87 Rb. It is conventionally expressed with respect to the lighter isotope of strontium, 86 Sr. Normally this relationship takes the form of an absolute ratio (in the region of 0.708) and so is not expressed in the conventional ’δ’ notation used in oxygen and carbon isotope studies. The Sr isotopic composition of seawater - and the rock derived from it - is controlled by the isotopic ratios in two terrestrial reservoirs: continental crust and the Earth’s upper mantle.
Continental crust supplies dissolved Sr to the oceans via weathering while the mantle supplies Sr to the oceans via hydrothermal activities at deep sea vents or ’black smokers’. Due to early isotopic separation at the time of the Earth’s formation these two reservoirs have developed distinctly different geochemical signatures, with continental crust having significantly higher Sr-87/Sr-86 ratios.
It is the interplay of the relative importance of these two sources that gives rise to the changing Sr isotopic composition of seawater and derived rocks through time. Because of the slow rate of change and the long residence time of well-mixed strontium in the ocean a single Sr isotope curve can be generated that is globally applicable. Or, to put it another way, a single measurement compared with a reference curve (eg Fig below) will place you accurately in time.
One thing that is immediately apparent is that, despite the slow rate of change of the curve Sr values are not time-unique, ie the value of about 0.709 that characterises the present day has also occurred in the Silurian and in the latest Cambrian. The point is that these repeat values are so widely separated in time that there is no danger of mixing them up. Fossils from the Holocene, Silurian and late Cambrian are all quite different and easily recognisable as such. Once you have identified what ’ballpark’ you are in, you can use the Sr isotope curve to refine your position in time.
Today many more Sr measurements are being generated and the reference curve is becoming progressively more refined. We are already at the stage where Sr isotope stratigraphy can be performed routinely and looks set to outstrip even carbon isotopes as a technique for orientating oneself in time, at least on longer timescales.
3. Recent trends
As technology has advanced, the discipline of chemostratigraphy has spread beyond just isotopes. Today, Inductively Coupled Atomic Emission Spectrometry (ICP-AES) and Inductively Coupled Plasma Mass Spectrometry (ICP-MS) are widely used to make rapid and simultaneous measurements of the abundance of up to 50 elements that can then be used to characterise a rock unit. This technique is increasingly applied in the petroleum exploration industry where the emphasis is on correlating local rocks (for example, tracing the lateral extent of hydrocarbon bearing sandstones). Future developments will see the use of portable laser spectrometers that can be used at the well site and which will remove the need for complex, laboratory based ICP-AES and ICP-MS analysis.
Sulphur, nitrogen and neodymium isotope chemostratigraphy is also advancing. Sulphur isotope variability is linked to the partitioning of sulphur between oxidised (sulphate) and reduced (sulphide) reservoirs. Bacterial reduction of sulphate preferentially incorporates the light isotope of sulphur, 32 S so that an increase in the rate of sulphide formation results in higher δ34S values.
Nitrogen isotopes in seawater and associated sediments reflect nitrate use and the rate of denitrification in seawater. Oxidation of dead plankton as it falls through the water column results in concentration of the heavy isotope, 15 N over the light isotope, 14 N, and an increase therefore in the δ15N ratio. Like elemental abundance chemostratigraphy, sulphur and nitrogen isotopes are susceptible to local effects and are therefore not likely to be useful on the global scale.
Neodymium isotopes in seawater and associated sediments, like strontium isotopes, reflect source rock composition and, as such, show promise as a potential tracer of global water mass movements.






No comments yet