X-ray structure of cannabinoid-regulating enzyme confirms computational predictions of drug binding and clarifies its mechanism of action
An enzyme that influences how we feel pain and is a potential target for treating anxiety has been crystallised and its structure solved by US scientists.1 The structure settles a long running question of how promising drug candidates bind in the active site, confirming computational predictions, and provides valuable information about the enzyme’s mechanism of action.
Raymond Stevens’s group from the Scripps Research Institute in La Jolla, California, solved the structure of fatty acid amide hydrolase (FAAH) when it is bound to URB597 - a potential drug candidate that blocks the enzyme’s activity. They also found a previously undiscovered water molecule in the active site, which gives some insight into why URB597 is active.
FAAH is responsible for breaking down natural endo-cannabinoids - the body’s signalling molecules that suppress pain by binding to cannabinoid CB1 and CB2 receptors, so blocking FAAH prolongs the body’s natural pain suppression. The structure has been solved before, but at lower resolution and never with URB597 bound.
Mauro Mileni, the team’s crystallographer, explains that FAAH cleaves an amide bond in the natural endo-cannabinoid substrates using the alcohol side chain of the amino acid serine to attack the amide. This leaves a flexible fatty acid ester covalently attached to the enzyme which must be removed before another molecule can be processed - and this is the job of the water molecule. ’But more rigid compounds like URB597 cannot move around to be in the right orientation for the water to attack,’ says Mileni, ’and they are joined to the enzyme by a carbamate rather than an ester - which is a much stronger bond - so they remain attached.’
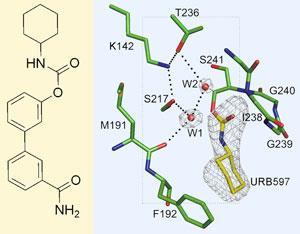
The crystal structure also confirms the orientation in which URB597 binds to FAAH. There are two channels either side of the active site, and knowing which end of the molecule binds in which channel would help medicinal chemists design more selective drugs, by designing them to fit more snugly. But this is more difficult than it sounds, says Mileni, and evidence for both orientations had been proposed.
Adrian Mulholland’s group at the University of Bristol, UK, used combined quantum mechanics and molecular mechanics (QM/MM) calculations to predict the orientation in 2008,2 and Mulholland is delighted to see that their prediction has been confirmed crystallographically. ’QM/MM methods have been regarded as quite esoteric, but this shows that they can be practically useful for drug development problems,’ he says.
Phillip Broadwith
References
et al, J. Mol. Bio., 2010, DOI:10.1016/j.jmb.2010.05.034
2. A Lodola et al, Chem. Commun., 2008, 214-216 (DOI:10.1039/b714136j)






No comments yet