Development of generic biological drugs is gathering momentum. But as Rajendrani Mukhopadhyay finds out, the welcome they are receiving looks very different in the EU and the US
Development of generic biological drugs is gathering momentum. But as Rajendrani Mukhopadhyay finds out, the welcome they are receiving looks very different in the EU and the US
Pharmaceutical companies and government regulatory agencies have been grappling with the issue of biosimilars for more than seven years on both sides of the Atlantic. Just as off-patent small molecule drugs are opened up to generic competition, companies are now beginning to make copies of biological therapies as their patents approach expiry.
But because biologics are much more complex than small molecule drugs, their duplicates are also complex. This complexity unleashes all kinds of regulatory and scientific issues, which the US and EU have tackled in strikingly different ways.

Biological drugs are generally protein based, although the class could be expanded to include other biomolecules, such as DNA or small interfering RNA (siRNA). Examples include peptide hormones such as insulin, erythropoietin (EPO) and human growth hormones, as well as more complex structures such as monoclonal antibodies. According to independent analyst firm Datamonitor, the biosimilars market was worth $243 million (?150 million) last year. By 2015, they predict the market will have grown to $3.7 billion.
Besides the lure of a share in this lucrative market, there is a social need for biosimilars. Generic versions will be slightly cheaper than the original biologics, which can cost patients, insurers or the NHS thousands of pounds a year for a single course. Janice Reichert, a pharmaceutical industry expert at the Tufts Center for the Study of Drug Development in Boston, US, illustrates the point with EPO. ’There’s a big market’ for the anaemia drug, she says. ’Reducing its cost by 20 per cent could result in significant savings to the healthcare system.’
Generic versions of small-molecule drugs undergo abbreviated approval processes with regulatory agencies, in which the generic manufacturer can rely on data from the original innovator drug to support its application. In theory, the same applies to biosimilars: expedited regulatory pathways
permit pharmaceutical companies to rely on what is already known about a drug to save time and resources, and avoid unnecessary duplication of human or animal testing. The problem is that, as the complexity of the drugs increases, it becomes increasingly hard to prove that the generic version is exactly the same as the original.
Laying ground rules
In 2004, the European Medicines Agency (EMA) established the regulatory framework for approving biosimilars in the EU. The EMA has issued general guidelines that apply to all biosimilars as well as specific guidelines for the specific classes of molecules, such as EPO, antibodies, and human growth hormone.1 In 2006, the human growth hormone, Omnitrope, was the first biosimilar approved in the EU. Today, 14 biosimilar products are authorised in Europe.

The US is only beginning to take action. The controversial healthcare reform legislation that President Barack Obama signed into law in March 2010 contained a provision for biosimilars, called the Biologics Price Competition and Innovation Act (BPCIA).2 Legal and pharmaceutical experts are now waiting breathlessly to see how the US Food and Drug Administration (FDA) will implement the legislation into a regulatory pathway.
Experts praise the EMA’s handling of biosimilars. ’You have to weigh the benefits of biosimilars to society against the potential for harming public safety if regulatory agencies are too quick to approve them,’ states Anurag Rathore, whose research at the Indian Institute of Technology, New Delhi, involves methods of manufacturing biopharmaceuticals. ’The EMA has done a great job in balancing the two. The FDA is more conservative, but they will have to pave the way’ because of the economic impact of biosimilars on healthcare.
Technical challenges
To understand the obstacles facing a company trying to produce generic biologics, it helps to compare them to small molecule pharmaceuticals. A typical small molecule drug contains around 20 to 100 or so atoms. Biologics can have anywhere from 190 to over 50,000 atoms. Small molecule drugs are chemically synthesised, easily purified by routine procedures, and well characterised. Biologics are usually produced as mixtures in genetically modified cells. The molecules in the mixture only differ slightly from one another, making them difficult to separate and characterise.
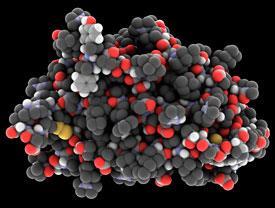
Some biologics are relatively simple, such as human growth hormone which only has 191 amino acids. Others are more complex, often being chemically modified and decorated after being translated from their DNA sequence. Monoclonal antibodies for example, comprise several polypeptide chains bristling with enormous tufts of sugar chains. The way a biosimilar is made - and the post-translational modifications on it - can differ from the innovator drug, as long as this doesn’t affect its therapeutic action. However, the generic version must have the exact same protein sequence as the molecule it’s supposed to replace.
In order to imitate an innovator biologic, a generics company has to study in detail what it looks like and how it works. But the biosimilar company is at a distinct disadvantage. ’The innovator is not going to give you the details of their analysis or production,’ says Pauline Rudd, head of the Oxford Glycobiology Laboratory at the National Institute for Bioprocessing Research and Training in Dublin, Ireland.
First, the innovator product’s protein sequence must be converted into the corresponding gene sequence. A cell line then has to be genetically manipulated with this sequence to produce the drug. Because the biosimilar company doesn’t know how the innovator company established the cell line to make the original drug, its cell line will most likely differ and affect the properties of the imitator drug.
The biosimilar company will then have to scale up their production route, all the while tracking parameters such as the cell growth media and the best time to harvest the proteins. Without clues about what the innovator company does, such as the specific chromatographic steps that are needed to get a properly purified product, the biosimilar company can be in for some unpleasant surprises and find their product doesn’t behave like the original, explains Alain Beck, head of physicochemistry at the Pierre Fabre Centre for Immunology in St. Julien-en-Genevois, France.
How similar is similar?
A biosimilar company then has to prove the efficacy and safety of their product to the regulatory agencies, which involves chemical and biochemical analysis, animal toxicology studies, and human clinical trials. Analytical and animal studies are always expected; in some cases, clinical studies can be skipped if the analytical and animal studies are convincing enough.
For the chemical analysis, multiple approaches - such as high performance liquid chromatography, capillary electrophoresis and mass spectrometry - are needed to piece together a biosimilar’s properties, including any post-translational modifications. Glycosylation (the pattern of sugar molecules tacked onto the peptide) causes the most headaches, because it makes for a diverse array of molecules. A single analytical approach just won’t cut it because it may be skewed towards picking up a certain molecular signature. As Michael Tarlov, the deputy chief of the biochemical science division at the US National Institute of Standards and Technology in Gaithersburg, Maryland explains: ’For glycosylation, the results can vary depending on what chromatographic method you use and certainly what mass spectrometric method you use.’
The cost and technical sophistication needed to make consistent copies of biologics that are safe and effective for patients makes the pursuit only possible for large companies, such as Sandoz (the generics arm of Swiss pharmaceutical firm Novartis) and Teva, the world’s largest generics company, based in Israel. ’Small start-up companies are unlikely to have the technology in place,’ says Rudd.
Mark McCamish is the head of global biopharmaceutical development at Sandoz (the company also produces novel biologic therapeutics). He explains that they track an innovator company’s product for years - with an arsenal of sophisticated analytical techniques - to understand a biologic’s characteristics. ’It takes us twice as long to develop a biosimilar as to produce a novel biologic,’ he claims.
Chemistry has to play a central role, says Rathore, if biosimilars are to have a sustainable future. ’What’s hurting biosimilars is our current over-reliance on clinical trials for assessment of safety and, to some extent, efficacy,’ he states. If chemists develop better immunogenicity assays, for example, that reduce the dependency on clinical trials, the field can quicken its pace. ’If we continue on the present path, large clinical trials will be needed to validate a drug’s safety and efficacy. The cost of these trials will hurt the economic feasibility of launching biosimilars.’
Making the switch
A branded medication and its generic version are considered to be interchangeable when a pharmacist can dispense the generic drug even when the branded medication is prescribed. Patients must also be able to switch between name brand and generic versions without adverse effects. Many small-molecule drugs are interchangeable, but opinions are sharply divided with biosimilars. The molecules’ complexity raises questions about how to determine whether the original and the copy are similar enough to be interchangeable. The fear is that if the molecules aren’t similar enough, the biosimilar may trigger devastating immune responses in patients.

The FDA calls interchangeability a ’higher standard’. Greg Peterson is a partner of the law firm of Bradley Arant Boult Cummings that has several offices across the US. He interprets ’higher standard’ to mean that the biosimilar company will likely be obliged to do clinical studies to prove its product can be swapped for the innovator product.
But McCamish says it’s a double standard. He and Reichert explain that, when biologics started to appear in the late 1980s, regulatory authorities realised how sensitive they were to manufacturing changes. Just a tweak of a chromatography column diameter would change the glycosylation pattern on a drug. So the regulatory authorities established guidelines. The innovator company has to compare the molecule before and after a manufacturing change to make sure it still fits the label; extra clinical trials are only required if regulatory authorities are unsure of the drug’s safety or efficacy after the change.
Sandoz surveyed several innovator biologics and found their composition substantially changed after tweaks in manufacturing processes. McCamish gives the example of Roche-Genentech’s Mabthera (rituximab), a drug for rheumatoid arthritis, non-Hodgkin’s lymphoma and chronic lymphocytic leukaemia. Despite a manufacturing change during a two-year period, which affected the molecule’s glycosylation pattern, the product was labelled and marketed the same way.
Two sides to the story
From the biosimilar companies’ viewpoint, the innovator’s manufacturing processes introduced changes to biologics that the FDA permitted to enter the market, often without extra clinical data. To tell biosimilar companies that they must go through clinical trials to gain approval for interchangeability, when the innovator usually doesn’t have to do the same after changing their processes, does not seem scientifically sound.
Joe Miletich is the senior vice president of research and development at Amgen, the Californian company that developed EPO and has several innovator biologics on the market. He says that the regulatory agencies should make clinical trials mandatory. ’It’s very difficult for me to understand how anyone can be certain biosimilars are going to perform the same way unless there is some degree of clinical comparison with the innovator drug,’ he says.
Rathore says the reality is that most biosimilar companies won’t know the therapeutics as intimately as the innovator companies. ’However, if both the innovator and the biosimilar producer have demonstrated the same level of process and product understanding, then the expectations for interchangeability should be the same for both,’ he says.
Because the EMA has acted promptly on biosimilars and issued guidelines, it’s clear what is expected from a biosimilar company to get their product approved in the EU. But the FDA is yet to set up its guidance, so the BPCIA is being analysed and interpreted to glean how the FDA may outline its regulations.
The EMA’s definition of a biosimilar is ’a product which has demonstrated similarity to the already authorised biological medicinal product in terms of quality, efficacy and safety through a comprehensive comparability exercise.’ The BPCIA states a pharmaceutical company must show that the biosimilar is ’highly similar’ to the reference product.
The term ’highly similar’ is what’s raising questions in the US. Given the challenges inherent in both producing and analysing biosimilars, Reichert and Peterson question how similarity will be assessed, and to what extent two molecules will be thought to be similar. This, in turn, makes it unclear how much and what types of data a biosimilar company will have to submit to the FDA.
So far, the FDA has been accepting biosimilar applications under its normal regulatory pathway. Based on its past actions, the experts anticipate that the FDA will use its judgment, like the EMA, and tackle biosimilars on a case by case basis rather than implement a rigid set of rules.
Great expectations
The field is globally ramping up as companies move more money to capitalise on expiring patents of innovator biologics. But everyone views the future as an uphill climb. ’I think there are enormous opportunities for all of us, but I do believe there is a bit of tension right now between the hope for a huge economic benefit and an under-appreciation for the level of care and complexity that’s going to be required,’ says Miletich.
McCamish sees the major hurdle to be education. He says innovator companies have passed off Asian versions of protein therapeutics as biosimilars to physicians and patients, with the message that biosimilars are markedly different from the originals and should be feared. ’These Asian products are not biosimilars,’ states McCamish - they have not been subjected to the same rigorous analysis and clinical demonstrations of comparability as approved biosimilar products.
Over the next five years, biosimilars will remain complicated and expensive to produce. But Beck remains optimistic: ’Even if it’s challenging, these products will come to market’ because the potential rewards are so high.
Rajendrani Mukhopadhyay is a freelance science journalist based in Washington, DC, US
Further Reading
1 EMA website on biosimilar regulations
2 FDA website, Implementation of the Biologics Price Competition and Innovation Act of 2009






No comments yet