
Diatomic nitrogen gas is inert because the triple bond requires a lot of energy to break. Metastable higher energy forms of nitrogen have been identified, notably N4, which was first produced by reducing the N4+ ion by Fulvio Cacace and colleagues at the Sapienza University of Rome, Italy, in 2002. N2 has a low melting point because there is no intrinsic charge separation within the molecule and thus little to attract one molecule to another. Barak Hirshberg and Robert Benny Gerber at the Hebrew University of Jerusalem and Anna Krylov of the University of Southern California in Los Angeles decided to investigate whether this was true for N4.
They used a computer algorithm to calculate the energy of an N4 crystal and optimise the structure to reduce this. The structure unexpectedly converged to a crystal of linear N8 molecules, with the two central atoms linked by a double bond and both single and triple bonds elsewhere in the molecule. Crucially, there is considerable charge separation, with some of the bonds having semi-ionic characteristics, which should cause attractive forces between the molecules allowing the molecular crystal to remain stable at higher temperatures. To check the result, the researchers repeated their calculations starting from a different structure. Each time, the optimisation procedure converged on the same solution.
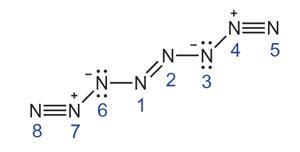
Artem Oganov, a computational materials theorist at Stony Brook University, US, says the result is ‘extremely interesting’, but cautions that a structure devised by modelling the progress of a reaction from a fixed starting point may not be the most stable or lowest energy possible solution. ‘They found one good metastable structure with rather good kinetic stability,’ he says. ‘It doesn't mean it's the most stable and it doesn't mean it's the lowest energy.’






No comments yet