Small molecule inhibitor of tuberculosis protein complex offers a new strategy to attack the disease
Researchers in the US have discovered that a class of small molecules can selectively disable a key protein complex in the tuberculosis bacterium and kill the organism, raising the possibility of new highly selective drugs to treat tuberculosis.
Carl Nathan, of Cornell University in New York, US, and his colleagues targeted a structure called the proteasome in Mycobacterium tuberculosis. This is a large complex of many proteins that plays a key role in maintaining the health of the cell by disposing of damaged proteins whose presence would otherwise cause the cell to die. If the proteasome is inactivated, unwanted proteins will accumulate and eventually poison the cell.
However, human cells also contain proteasomes and these are structurally similar to those in M. tuberculosis. So far no-one has managed to find a compound that will interfere with one and not the other. Nathan’s team screened a library of 20,000 compounds against the bacterial proteasome and found that two were potent inhibitors - both oxathiazol-2-ones, which have a five-membered ring containing nitrogen, sulphur and oxygen. Surprisingly and importantly, the compounds did not significantly inhibit the human proteasome.
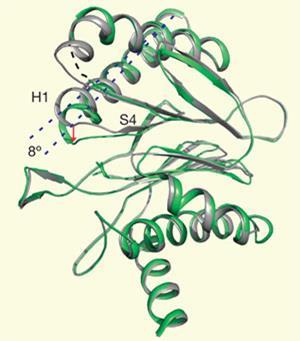
To understand why the molecule behaved differently in two protein complexes that have a similar active site, the team examined the crystal structure of the proteasome interacting with the inhibitor. In the proteasome active site the oxathiazol-2-one covalently attacks an OH group of a threonine residue, forming a carbonated or carbothionated intermediate.
In the tuberculosis proteasome, this reaction results in a chemically stable product - essentially fouling up the active site and crippling the protein’s function. In the human proteasome, however, the intermediate becomes hydrolysed and the activity of the enzyme is restored. ’It chews it up and spits it out,’ says Nathan. It seems that this difference is caused by a conformational shift in the tuberculosis proteasome, where a structural loop of the protein swings across the active site, preventing water from entering it - hence no hydrolysis is possible. A similar ’loop shift’ does not happen in the human proteasome, allowing the hydrolysis to occur. The precise reasons for this shift are not clear, but it appears to confer a crucial difference that could be exploited in drug development.
Geoff Coxon of Strathclyde University in the UK is director of medicinal chemistry for a newly formed body called Tuberculosis Drug Discovery UK. He says that the new work is important as it represents the basis to initiate drug discovery programmes selectively targeting mycobacterial proteasomes. ’In previous efforts this has been difficult as we have not been able to understand how the proteasome from M. tuberculosis can be inhibited selectively over the human versions,’ explains Coxon. ’This has made it difficult to justify this as a drug discovery strategy as any new drugs that would have been produced would potentially be lethal to the patient. This work now provides a way to avoid this problem.’
Simon Hadlington
References
G Lin et al, Nature, 2009, DOI: 10.1038/nature08357






No comments yet