A simple way to insert fluorine into organic compounds could help chemists manufacture improved drugs
US researchers have discovered a simple, low-cost way to add fluorine atoms to heteroaromatic rings. Inserting fluorine into molecules that are drug candidates can give the compounds many improved properties, such as increased stability or improved interaction with target proteins.
Trifluoromethylation of heteroaromatic compounds is a key reaction in many organic syntheses, and is widely used in the development of new pharmaceuticals and agrochemicals. ’One procedure involves the gas CF3I which is difficult to measure and medicinal chemists do not like using it,’ says Phil Baran, of the Scripps Research Institute in California, who led the new research.
Another common method is to use anhydrous hydrogen fluoride as the fluorinating agent, explains Graham Sandford, who researches organofluorine chemistry at the University of Durham, UK. ’For discovery chemists, using anhydrous hydrogen fluoride is not very convenient to say the least and direct, efficient trifluoromethylation methods have long been sought.’
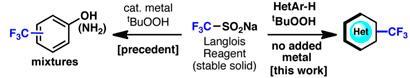
Baran’s team set about searching for a simpler method for trifluoromethylation of heterocycles. The team tested scores of reagents in more than 500 reaction systems for the trifluoromethylation of a test compound, 4-t-butylpyridine. One reagent achieved the desired result: sodium trifluoromethanesulfinate, CF3SO2Na, a stable, inexpensive solid also known as Langlois reagent.
The CF3 radical is generated through induced decomposition of the reagent. But, Baran says: ’The addition of the electron-deficient radical to electron-deficient heterocycles was counterintuitive and a surprise.’ No metal catalyst was necessary. The radical attaches to the heterocycle at the most innately reactive C-H bonds on the substrate, and the point of attack is largely predictable. A degree of control can be exerted by choice of solvent, Baran says. The method works on complex heteroaromatic molecules and can tolerate the presence of multiple functional groups.
The new technique, in principle, should make it easier for pharmaceutical companies to modify and improve specific drug compounds of interest. ’It also means that these companies can expand the existing compound libraries they use for drug-discovery screening by making trifluoromethylated versions of these compounds quickly and easily,’ says Baran.
’Medicinal chemistry exploration of pharmacophore space and ligand interactions benefits tremendously from methods that introduce substituents late in syntheses,’ says Jotham Coe, a medicinal chemist at Pfizer. ’Such reactions are best when versatile and reliable enough to work in conjunction with variable substitution within target molecule classes. The Baran chemistry addresses these shortcomings economically, providing options never before available.’
The work represents ’a significant advance’, Sandford says, ’especially since highly functional heterocycles and natural products may be trifluoromethylated regioselectively.’
Simon Hadlington
Interesting? Spread the word using the ’tools’ menu on the left.
References
Y Ji 10.1073/pnas.1109059108







No comments yet