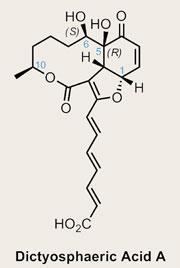
Dictyosphaeric acid A
Dictyosphaeric acid A
The total synthesis of macrolide targets is now a relatively mature field, with many well-explored methods for their synthesis established. Any synthesis that bucks these trends grabs attention, with a recent publication of dictyosphaeric acid A by Richard Taylor’s team at the University of York, UK, a case in point.1
Isolated in 2004, the target is a particularly appealing one, with both an interesting structure and an attractive biological profile.2 The latter includes antibacterial activity against methicillin-resistant Staphylococcus aureus(MRSA), with the former a tricyclic macrolide containing four contiguous stereocentres - so there’s something for everybody in this molecule. The structure-activity relationship is made all the more intriguing when the related dictyosphaeric acid B - containing one further hydroxyl group at the expense of an olefin - is inactive.
Given these facts, it’s quite surprising that this is actually its first synthesis. Further, the structural assignment of the C-6 hydroxyl group at isolation was somewhat tentative. The original assignment suggested this group is 5S,6S configured, but Taylor’s reading of the NMR suggested that 5S,6R might also fit the published data.
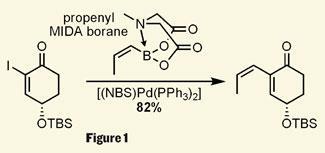
Taylor’s chemistry caught my eye from the very beginning of the synthesis, with a rather special Suzuki coupling. This palladium mediated reaction is now thirty years old, and each year brings new refinements and modifications - but I’m clearly falling behind on my reading, as the exotic looking boronic acid is actually commercially available - propenyl N-methyliminodiacetyl (MIDA) boronate, developed by Martin Burke of the University of Illinois at Urbana Champaign, US, (see Chemistry World, December 2008, p20).3 The MIDA cage built around the vinyl borane provides stability, slowly releasing the active boronic acid under the reaction conditions. The catalyst is also pretty special - developed by the team for particularly challenging substrates, this organometallic is refreshingly simple to make from common palladium precursors and N-bromosuccinimide (NBS, figure 1).4
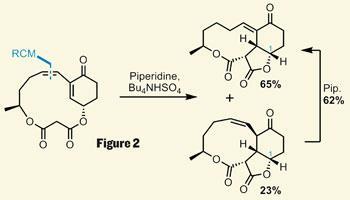
In testament to a concise synthesis, the group had completed the larger ring only four steps later, with a ring closing metathesis (RCM) reaction. Using a little base, they performed a very neat intramolecular Michael addition - deprotonating the malonyl group, which then added to cyclohexenone, creating two stereocentres. Unfortunately, nearly a third of the material was left as an isomer of the desired product, but a little more base rectified the situation, migrating the double bond into conjugation (figure 2).
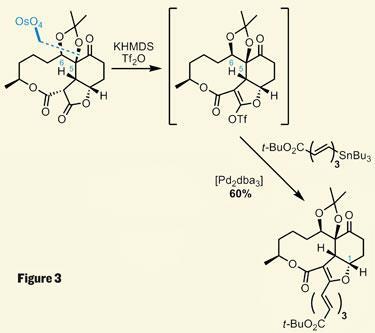
Positioning of this alkene was key, as it allowed a substrate-controlled dihydroxylation to impart both of the hydroxyl groups with the desired stereochemistry. Control of regioselectivity was then vital, as the group wished to select between the two malonyl carbonyls to form an enol triflate. Using a strong base and triflic anhydride was gratifyingly selective for the smaller ring carbonyl, providing the group with a handle for more metal-catalysed cross coupling chemistry. A more conventional palladium catalyst mediated this Stille coupling, working with a trienylstannane that looks quite sensitive to my paranoid eye (figure 3).
Two more steps completed the molecule - a Saegusa-Ito oxidation provided the enone required in the target, and a little acid both removed the acetonide protecting group and unmasked the side chain acid. With the molecule complete, rigorous analysis of its NMR and optical activity compared favourably with the original isolate - confirming the structure and stereochemistry. The result agreed with Taylor’s reassignment, concluding a tour-de-force of macrolide chemistry.
Paul Docherty is a science writer and blogger based in London, UK
References
1 A R Burns et al, Angew. Chem. Int. Ed., 2010, DOI: 10.1002/anie.201002416
2 C M Ireland et al, J. Nat. Prod., 2004, 67, 1396
3 D M Knapp, E P Gillis and M D Burke, J. Am. Chem. Soc., 2007, 129, 6716
4 M J Burns et al, Org. Lett ., 2007, 9, 5397












No comments yet