Kendomycin
Kendomycin
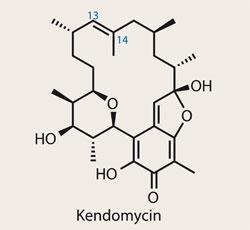
Although antiviral compounds are currently topping the popular science press (it’s good to see the products of my day-job so in demand), the pursuit of novel antibiotics is never far from the news. With potent bacteria-beating activity,1 it’s no surprise that kendomycin has recently grabbed quite a bit of attention.2,3,4 Johann Mulzer of the University of Vienna in Austria proved his interest in its total synthesis sometime ago, publishing his initial synthetic studies back in 2003, but now brings us not one but two new syntheses.5
Kendomycin is a macrocycle, and the problems with making such structures tend to come when assembling the larger fragments toward the end of the synthesis - hence Mulzer’s two-pronged approach. While synthesis of the smaller fragments is relatively easy, as much of the chemistry is well known, fitting these fragments together is a trickier prospect as each target is idiosyncratic - and seemingly perfect retrosyntheses fall apart. In the case of kendomycin, closing the macrocycle with metathesis about the C-13,14 olefin looks perfect - but doesn’t work. So Mulzer considered a couple of intriguing alternatives.
Molecular backflips
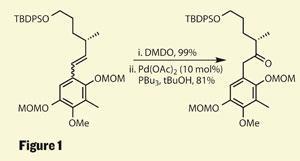
A theme throughout this synthesis is rearrangement chemistry - an early example comes as Mulzer makes the molecule’s right hand fragment. This portion was put together by an unselective Negishi coupling, clipping an aryl ring to a vinyl iodide to form a mixture of olefin isomers, which Mulzer then epoxidised. A bit of palladium prompted rearrangement of the epoxides to give the desired ketone in a cracking yield (figure 1).
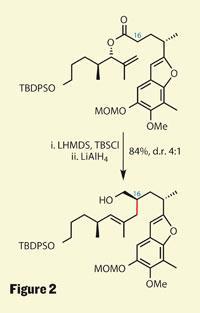
While that rearrangement chemistry was new to me, a far more established reaction was used later to construct a challenging stereocentre. The Claisen rearrangement, and its many derivatives (the Johnson-Claisen is my particular favourite), have been used by synthetic chemists for almost a century, and its appeal isn’t diminishing. This partly stems from their generally predictable stereochemical outcome - and the Ireland variant is particularly useful in this regard. Using a little base, this reaction can transform a stereodefined ester into a new stereodefined C-C bond. Mulzer attempted a particularly complex example of this reaction as a key C-C bonding step, and was rewarded with both a high yield and useful selectivity (figure 2). Even better, the precursor to this reaction was a simple esterification, proving that tethering two fragments together and then rearranging to provide the desired linkage is a powerful strategy.
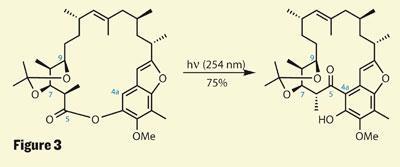
The final rearrangement is somewhat similar, in that a comparatively simple tether is rearranged to provide a key C-C linkage - but the manner of the reaction is quite different. The Fries rearrangement (again due its long-service badge at just over a century) is quite different in its mechanism, and probably involves fragmentation of the macrocyclic ring followed by prompt re-formation. It also comes in two distinctly different flavours - Lewis acid mediated and the so-called photo-Fries. This is handy, as the idea of throwing a pokey Lewis acid at Mulzer’s advanced intermediate brings me out in a cold sweat (I can already see the protecting groups getting chewed-off). Using photons - a cheap and abundant reagent - triggers a radical pathway, and a great yield of the desired macrocycle, which is just a few functional group conversions away from the natural product itself (figure 3).
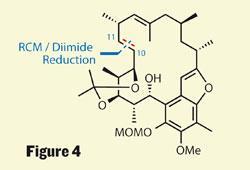
Mulzer’s other strategy was to perform a ring-closing metathesis to create the macrocycle. This approach, as I mentioned earlier, was somewhat traumatic, as a C-13,14 linkage was unworkable. The group persevered, however, and attempted various metatheses about the C-9,10 and C-19,20 positions, but were again denied success. However, the C-10,11 bond appeared more suitable, returning a respectable 62 per cent yield, but leaving an unwanted double bond in the ring and hence a rather tricky reduction (figure 4). With two other olefins present in the molecule, selectivity was key, and by using the slightly exotic diimide reagent the group did the job. An impressive combination of retrosynthesis and perseverance.
Paul Docherty is a medicinal chemist based in London, UK
References
1 H B Bode and A Zeeck, J. Chem. Soc. Perkin Trans. 1, 2000, 323 (DOI: 10.1039/a908387a)
2 A B Smith III, E F Mesaros and E Meyer, J. Am. Chem. Soc., 2006, 128, 5292 (DOI: 10.1021/ja060369+)
3 J T Lowe and J S Panek, Org. Lett., 2008, 10, 3813 (DOI: 10.1021/ol801499s)
4 K B Bahnck and S D Rychnovsky, J. Am. Chem. Soc., 2008, 130, 13 177 (DOI: 10.1021/ja805187p)
5 T Magauer, H J Martin and J Mulzer, Angew. Chem. Int. Ed., 2009, DOI: 10.1002/anie.200900522












No comments yet