Pseudolaric acid B
Pseudolaric acid B
Regular readers of this column’s online incarnation will have noticed that this is the second appearance for this particular synthesis. Its first showing was in a particularly short JACS communication last year, which simply didn’t do justice to the amazing breadth of chemistry Stanford University’s Barry Trost had employed. However, good things come to those that wait - especially in the lab - and we now have an engrossing full paper, and a lot of chemistry.1
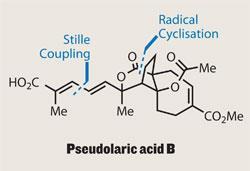
There’s a load of biology too, with a hugely impressive activity profile accompanying that intriguing structure. This tricyclic beasty inhibits angiogenesis, causes apoptosis in human melanoma cells, and inhibits the polymerisation of tubulin, among other roles. But how to build the tricycle?
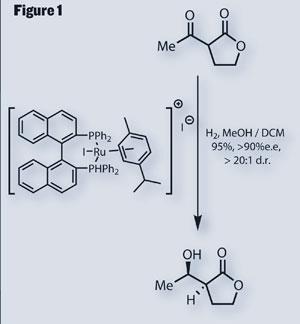
Not to get too far ahead of ourselves, we’ll start at the beginning, with an impressive asymmetric hydrogenation to install two key stereocentres (figure 1). This reaction, developed by Hidemasa Takaya, results in a fantastic yield, enantiomeric excess and distereomeric ratio - everything you could ask for.2
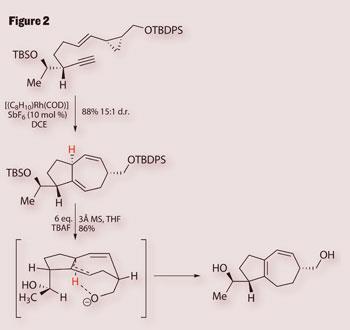
With the stereochemistry set, Trost’s team quickly extended the structure, readying it for a rhodium-catalysed [5+2] cycloaddition which give the natural product’s 7, 5-fused ring system in good yield (figure 2). The team used a catalysis developed just down the hall, in Paul Wender’s Stanford lab.3 Initially, they’d hoped to use their own chemistry, using a not-entirely dissimilar ruthenium catalyst, but this gave a mixture of isomeric products.4
Happy accidents
The next piece of chemistry - rather more low-tech - saw the group trying to isomerise a double bond in the resulting cycloheptadiene, to bring the diene moiety into conjugation. Normally, this is the kind of reaction that would be all too easy to trigger accidentally, so the group must have been surprised that it was so hard to find reaction conditions that worked. However, as luck would have it, the isomerisation was started simply by using dry TBAF - which the team had actually added to take off the silicon protecting group (figure 2). It seems that removing the protecting groups gave a ’naked’ alkoxide that sits in just the right conformation to nab the proton (shown in red) that triggers isomerisation.
The team were struck by a second happy accident just a couple of steps later. Epoxidising the now-conjugated diene using m CPBA resulted in clean epoxidation of the desired olefin, but as a mixture of diastereoisomers (figure 3). Although the ratio was distinctly in favour of the isomer the team were after, they found - as is often the case - that separating the two was difficult. Like many chemists before them, the team opted to ’take the material through’, hoping that the mixture would be separable a few steps further on - a strategy that sometimes works, and sometimes does not. So in the next step - a base-triggered rearrangement to pop open the epoxide - the group must have been astonished to see that only one of the isomers reacted, leaving the unwanted epoxide behind. They would then have found the separation relatively easy, but the mechanism a bit more complex.
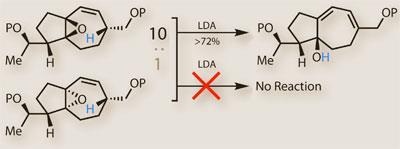
Trost suggests that the base - LDA - can deprotonate the allylic proton (shown in blue) only when coordinated by the epoxide. In the desired case, both are relatively close together on the same face of the molecule, so the reaction works well. In the case of the impurity, the opposite is true, and the epoxide resists. That must’ve been a good day in the lab.
Several steps later, the group were ready to introduce the unsaturated sidechain, by attaching an acetylene onto a ketone. Although these reactions probably hark back a century, I was again back in class, learning that cerium acetylides are alternatives to other more frequently employed lithium or Grignard reagents. But why bother - other than for the kudos of using a lanthanide-organometallic? In this case, because the usual suspects wouldn’t play ball. It’s not a ’new’ reaction, but learning new and alternative reagents is an essential process for the intrepid synthesiser.
And that’s one of the best aspects of this paper - novel reagents and interesting mechanisms. New stuff’s fun, isn’t it?
Paul Docherty is a medicinal chemist based in London, UK
References
et al, Chem. Commun., 2002, 2480
4 K C Nicolaou et al, Angew. Chem. Int. Ed., 2005, 44, 3874
5 G Guella et al, Angew. Chem. Int. Ed., 1999, 38, 1134












No comments yet